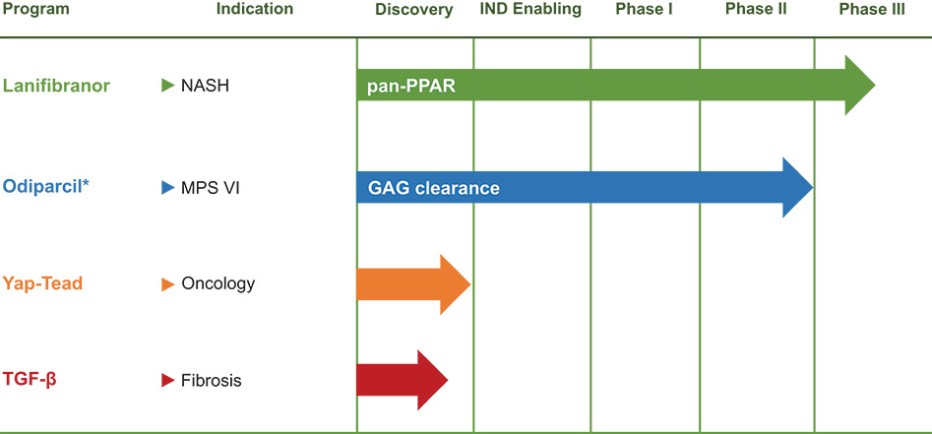
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws, regulations or permitting requirements. These current or future laws, regulations and permitting requirements may impair our research, development or production efforts. Failure to comply with these laws, regulations and permitting requirements also may result in substantial fines, penalties or other sanctions.
We are subject to stringent and changing U.S. and foreign laws, regulations, and rules, contractual obligations, industry standards, policies and other obligations related to privacy, data security, and data protection. Our actual or perceived failure to comply with such obligations could lead to regulatory investigations or actions; litigation (including class claims) and mass arbitration demands; fines and penalties; disruptions of our business operations; reputational harm; loss of revenue or profits; loss of customers or sales; and other adverse business consequences.
In the ordinary course of business, we collect, receive, store, process, generate, use, transfer, disclose, make accessible, protect, secure, dispose of, transmit, and share (collectively, processing) proprietary, confidential and sensitive data, including personal data (such as health-related data), proprietary and confidential business data, trade secrets, intellectual property, data we collect about trial participants in connection with clinical trials, and sensitive third-party data (collectively, sensitive information). Our data processing activities subject us to numerous data privacy, data security, and data protection obligations, such as various laws, regulations, guidance, industry standards, external and internal privacy and security policies, contracts, and other obligations relating to data privacy and security.
In the United States, federal, state, and local governments have enacted numerous data privacy and security laws, including federal health information privacy laws, state data breach notification laws, state health information privacy laws, consumer protection laws (e.g., Section 5 of the Federal Trade Commission Act) and other similar laws (e.g., wiretapping laws). In the past few years, numerous U.S. states—including California, Virginia, Colorado, Connecticut, and Utah—have enacted comprehensive privacy laws that impose certain obligations on covered businesses, including providing specific disclosures in privacy notices and affording residents with certain rights concerning their personal data. As applicable, such rights may include the right to access, correct, or delete certain personal data, and to opt-out of certain data processing activities, such as targeted advertising, profiling, and automated decision-making. The exercise of these rights may impact our business and ability to provide our products and services. Certain states also impose stricter requirements for processing certain personal data, including sensitive information, such as conducting data privacy impact assessments. These state laws allow for statutory fines for noncompliance. For example, the California Consumer Privacy Act of 2018, as amended by the California Privacy Rights Act of 2020, or CPRA, or collectively the CCPA, applies to personal data of consumers, business representatives, and employees who are California residents, and requires businesses to provide specific disclosures in privacy notices and honor requests of such individuals to exercise certain privacy rights. The CCPA provides for fines for noncompliance (up to $7,500 per intentional violation) and allows private litigants affected by certain data breaches to recover significant statutory damages. Similar comprehensive privacy laws have been passed or are being considered in several other states, as well as at the federal and local levels, and we expect more states to pass similar laws in the future. Although the CCPA and other states exempt some data processed in the context of clinical trials as well as protected health information under the Health Insurance Portability and Accountability Act of 1996, or HIPAA, these developments may further complicate compliance efforts and may increase legal risk and compliance costs for us and the third parties upon whom we rely.
In addition, we obtain certain information from third parties (including research institutions from which we obtain clinical trial data) that is subject to privacy and security requirements under HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH. HIPAA imposes specific requirements relating to the privacy, security, and transmission of individually identifiable health information. See “—Our current and future operations are subject to applicable fraud and abuse, transparency, government price reporting, and other healthcare laws and regulations. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.”
Outside the United States, an increasing number of laws, regulations, and industry standards govern data privacy and security. For example, the European Union’s General Data Protection Regulation (“EU GDPR”), the United Kingdom’s GDPR (“UK GDPR”), Brazil’s General Data Protection Law (Lei Geral de Proteção de Dados Pessoais, or “LGPD”) (Law No. 13,709/2018), and China’s Personal Information Protection Law (“PIPL”) impose strict requirements for processing personal data.