UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form
OR
For
the fiscal year ended
OR
Date of event requiring this shell company report ________________
For the transition period from _______________________________ to _______________________________
Commission
File No.
(Exact name of Registrant as specified in its charter)
N/A
(Translation of the Registrant’s name into English)
State
of
(Jurisdiction of incorporation or organization)
(Address of principal executive offices)
President and Chief Executive Officer
E-mail:
Tel:
Fax: +972.3.693.8447
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act.
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||
Securities registered or to be registered pursuant to Section 12(g) of the Act.
| N/A | ||
| (Title of each class) |
Securities registered or to be registered pursuant to Section 15(d) of the Act.
| N/A | ||
| (Title of each class) |
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report (December 31, 2021): ordinary shares are outstanding
Indicate
by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
If
this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section
13 or 15(d) of the Securities Exchange Act of 1934. Yes ☐
Note – Checking the box above will not relieve any registrant required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 from their obligations under those Sections.
Indicate by check
mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934
during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject
to such filing requirements for the past 90 days.
Indicate by check
mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of
Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required
to submit and post such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or an emerging growth company. See definition of “large accelerated filer,” “accelerated filer,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ☐ | Accelerated filer ☐ | ||
| Emerging
growth company |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards † provided pursuant to Section 13(a) of the Securities Act. ☐
† The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
Indicate
by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness
of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered
public accounting firm that prepared or issued its audit report.
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
| International Financial Reporting Standards | Other ☐ | |
| as issued by the International Accounting Standards Board ☐ |
If “Other” has been checked in response to the previous question indicate by check mark which financial statement item the Registrant has elected to follow: Item 17 ☐ Item 18 ☐
If
this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange
Act). Yes ☐
TABLE OF CONTENTS
| 2 |
ABOUT THIS ANNUAL REPORT
All references to “we,” “us,” “our,” “the Company” and “our Company”, in this Annual Report on Form 20-F, or our annual report, are to Galmed Pharmaceuticals Ltd. and its subsidiaries, unless the context otherwise requires. All references to Aramchol mean Aramchol acid or Aramchol meglumine (salt), unless the context otherwise requires. All references to “shares” or “ordinary shares” are to our ordinary shares, NIS 0.01 nominal par value per share. All references to “Israel” are to the State of Israel. “U.S. GAAP” means the generally accepted accounting principles of the United States. Unless otherwise stated, all of our financial information presented in this annual report has been prepared in accordance with U.S. GAAP. Any discrepancies in any table between totals and sums of the amounts listed are due to rounding. Unless otherwise indicated, or the context otherwise requires, references in this annual report to financial and operational data for a particular year refer to the fiscal year of our company ended December 31 of that year.
Our reporting currency and financial currency is the U.S. dollar. In this annual report, “NIS” means New Israeli Shekel, and “$,” “US$” and “U.S. dollars” mean United States dollars.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This annual report contains forward-looking statements about our expectations, beliefs or intentions regarding, among other things, our product development efforts, business, financial condition, results of operations, strategies or prospects. In addition, from time to time, we or our representatives have made or may make forward-looking statements, orally or in writing. Forward-looking statements can be identified by the use of forward-looking words such as “believe,” “expect,” “intend,” “plan,” “may,” “should,” “anticipate,” “could,” “might,” “seek,” “target,” “will,” “project,” “forecast,” “continue” or their negatives or variations of these words or other comparable words or by the fact that these statements do not relate strictly to historical matters. These forward-looking statements may be included in, among other things, various filings made by us with the U.S. Securities and Exchange Commission, or the SEC, press releases or oral statements made by or with the approval of one of our authorized executive officers. Forward-looking statements relate to anticipated or expected events, activities, trends or results as of the date they are made. Because forward-looking statements relate to matters that have not yet occurred, these statements are inherently subject to risks and uncertainties that could cause our actual results to differ materially from any future results expressed or implied by the forward-looking statements. Many factors could cause our actual activities or results to differ materially from the activities and results anticipated in forward-looking statements, including, but not limited to, the factors summarized below:
| ● | the timing and cost of our pivotal Phase 3 ARMOR trial, or the ARMOR Study, for our product candidates, Aramchol and Amilo-5MER, or for any other pre-clinical or clinical trials; | |
| ● | completion and receiving favorable results of the ARMOR Study for Aramchol or any other pre-clinical or clinical trial; | |
| ● | the impact of the COVID-19 pandemic on our operations; | |
| ● | regulatory action with respect to Aramchol or any other product candidate by the U.S. Food and Drug Administration, or the FDA, or the European Medicines Authority, or EMA, or the Medicines and Healthcare Products Regulatory Agency, or the MHRA, including but not limited to acceptance of an application for marketing authorization, review and approval of such application, and, if approved, the scope of the approved indication and labeling; | |
| ● | the commercial launch and future sales of Aramchol and any future product candidates; | |
| ● | our ability to comply with all applicable post-market regulatory requirements for Aramchol, Amilo-5MER or any other product candidate in the countries in which we seek to market the product; |
| 3 |
| ● | our ability to achieve favorable pricing for Aramchol, Amilo-5MER or any other product candidate; | |
| ● | our expectations regarding the commercial market for non-alcoholic steato-hepatitis, or NASH, in patients or any other targeted indication; | |
| ● | third-party payor reimbursement for Aramchol, Amilo-5MER or any other product candidate; | |
| ● | our estimates regarding anticipated capital requirements and our needs for additional financing; | |
| ● | market adoption of Aramchol or any other product candidate by physicians and patients; | |
| ● | the timing, cost or other aspects of the commercial launch of Aramchol or any other product candidate; | |
| ● | our ability to obtain and maintain adequate protection of our intellectual property; | |
| ● | the possibility that we may face third-party claims of intellectual property infringement; | |
| ● | ability to manufacture our product candidates in commercial quantities, at an adequate quality or at an acceptable cost; | |
| ● | our ability to establish adequate sales, marketing and distribution channels; | |
| ● | intense competition in our industry, with competitors having substantially greater financial, technological, research and development, regulatory and clinical, manufacturing, marketing and sales, distribution and personnel resources than we do; | |
| ● | the development and approval of the use of Aramchol or any other product candidate for additional indications or in combination therapy; and | |
| ● | our expectations regarding licensing, acquisitions and strategic operations. |
We believe these forward-looking statements are reasonable; however, these statements are only current predictions and are subject to known and unknown risks, uncertainties and other factors that may cause our or our industry’s actual results, levels of activity, performance or achievements to be materially different from those anticipated by the forward-looking statements. We discuss many of these risks in this annual report in greater detail under the heading “Risk Factors” and elsewhere in this annual report. Given these uncertainties, you should not rely upon forward-looking statements as predictions of future events.
All forward-looking statements attributable to us or persons acting on our behalf speak only as of the date hereof and are expressly qualified in their entirety by the cautionary statements included in this annual report. We undertake no obligations to update or revise forward-looking statements to reflect events or circumstances that arise after the date made or to reflect the occurrence of unanticipated events. In evaluating forward-looking statements, you should consider these risks and uncertainties.
| 4 |
EXPLANATORY NOTE
Market data and certain industry data and forecasts used throughout this annual report were obtained from internal company surveys, market research, consultant surveys commissioned by the Company, publicly available information, reports of governmental agencies and industry publications and surveys. Industry surveys, publications, consultant surveys commissioned by the Company and forecasts generally state that the information contained therein has been obtained from sources believed to be reliable. However, this information may prove to be inaccurate because of the method by which some of the data for the estimates is obtained or because this information cannot always be verified with complete certainty due to the limits on the availability and reliability of raw data, the voluntary nature of the data gathering process and other limitations and uncertainties. As a result, the market and industry data and forecasts included or incorporated by reference in this annual report, and estimates and beliefs based on that data, may not be reliable. We have relied on certain data from third-party sources, including internal surveys, industry forecasts and market research, which we believe to be reliable based on our management’s knowledge of the industry. However, we have not ascertained the underlying economic assumptions relied upon therein. Forecasts are particularly likely to be inaccurate, especially over long periods of time. In addition, we do not necessarily know what assumptions regarding general economic growth were used in preparing the forecasts we cite. Statements as to our market position are based to the best of our knowledge on the most currently available data. While we are not aware of any misstatements regarding the industry data presented in this annual report, our estimates involve risks and uncertainties and are subject to change based on various factors, including those discussed under the heading “Risk Factors” in this annual report.
PART I
ITEM 1. Identity of Directors, Senior Management and Advisers.
Not applicable.
ITEM 2. Offer Statistics and Expected Timetable.
Not applicable.
ITEM 3. Key Information.
A. [Reserved.]
B. Capitalization and Indebtedness.
Not applicable.
C. Reasons for the Offer and Use of Proceeds.
Not applicable.
D. Risk Factors.
Summary of Risk Factors
An investment in our ordinary shares is subject to a number of risks. The following summarizes some, but not all, of these risks. Please carefully consider all of the information discussed in “Item 3. Key Information—D. Risk Factors” in this annual report for a more thorough description of these and other risks.
Risks Related to Our Financial Position and Capital Requirements
| ● | We are a clinical-stage biopharmaceutical company with a history of operating losses. We expect to incur significant additional losses in the future and may never be profitable. | |
| ● | We have not yet commercialized any products and we may never be able to do so, and even if we do, the products may not gain market acceptance. |
| 5 |
| ● | We will need substantial, additional capital in the future. If additional capital is not available, we will have to delay, reduce or cease operations. | |
| ● | We are unable to estimate our long-term capital requirements due to uncertainties associated with the development and commercialization of Aramchol, Amilo-5MER or any other product candidate. If we fail to obtain necessary funds for our operations, we will be unable to develop and commercialize Aramchol, Amilo-5MER or any other product candidate. |
Risks Related to Our Business, Industry and Regulatory Requirements
| ● | The clinical trial process is complex and expensive, and commencement and completion of clinical trials can be delayed or prevented for a number of reasons. | |
| ● | We are developing Aramchol for the treatment of NASH, an indication for which there are no approved products, and there is significant uncertainty regarding the regulatory approval process. This makes it difficult to predict the timing and costs of the clinical development of Aramchol for the treatment of NASH. | |
| ● | We depend largely on the success of our lead product candidate, Aramchol, and we may not obtain regulatory approval of Aramchol. | |
| ● | Commencement of our ARMOR Study in jurisdictions outside the United States is subject to acceptance of the foreign equivalent of our IND by regulatory authorities. | |
| ● | We may be forced to abandon development of Aramchol or any other product candidate which would have a material adverse effect on our business and may force us to cease operations. | |
| ● | The lack of a reliable non-invasive method for the diagnosis of NASH is likely to present a major challenge to Aramchol’s market penetration, if ever commercialized. | |
| ● | Our Amilo-5MER program is being conducted under a license agreement with Yissum Research Development Company of the Hebrew University of Jerusalem, or Yissum. | |
| ● | If we acquire or in-license additional technologies or product candidates, we may incur significant, incremental expenses, may have integration difficulties and may experience other risks that could harm our business and results of operations. | |
| ● | Changes in regulatory requirements and guidance or unanticipated events during our clinical trials may occur, which may result in necessary changes to clinical trial protocols, which could result in increased costs to us, delay our development timeline or reduce the likelihood of successful completion of our clinical trials. | |
| ● | We manage our business through a small number of senior executive officers. We depend on them even more than similarly- situated companies. | |
| ● | Our business is subject to risks arising from epidemic diseases, such as the recent COVID-19 pandemic, which has impacted and could continue to impact our business. |
| 6 |
Risks Related to Our Reliance on Third Parties
| ● | We have no manufacturing capacity and anticipate reliance on third-party manufacturers for Aramchol, Amilo-5MER or any other product candidate. | |
| ● | Any collaboration arrangements that we may enter into in the future may not be successful, which could adversely affect our ability to develop and commercialize our current and potential future product candidates. | |
| ● | We depend on third parties to conduct our clinical trials. |
Risks Related to Our Intellectual Property
| ● | The failure to obtain or maintain patents, licensing agreements and other intellectual property rights that are sufficiently broad and protective could impact our ability to compete effectively. | |
| ● | Our potential development of Aramchol meglumine may not result in improved bioavailability compared to the existing form of Aramchol. Furthermore, although we have pending patent applications and granted patent covering Aramchol meglumine in development, there is no assurance that we will receive any patents for them, and even if we receive one or more patents for our Aramchol meglumine in development, they may be of little or no commercial value. |
Risks Related to Ownership of Our Ordinary Shares
| ● | We are currently operating in a period of economic uncertainty and capital markets disruption, which has been significantly impacted by geopolitical instability due to the ongoing military conflict between Russia and Ukraine. | |
| ● | The market price of our ordinary shares is volatile and you may sustain a complete loss of your investment. | |
| ● | Our President and Chief Executive Officer, along with our principal shareholders, beneficially own approximately 17.1% of our outstanding ordinary shares, as of April 15, 2022. Therefore, our principal shareholders will be able to exert significant control over matters submitted to our shareholders for approval. | |
| ● | Our U.S. shareholders may suffer adverse tax consequences due to our classification as a passive foreign investment company. |
Risks Related to Israeli Law and Our Operations in Israel
| ● | Our headquarters and other significant operations are located in Israel and, therefore, our results may be adversely affected by political, economic and military instability in Israel. | |
| ● | Provisions of Israeli law and our articles of association, or Articles, may delay, prevent or otherwise impede a merger with, or an acquisition of, our company, which could prevent a change of control, even when the terms of such a transaction are favorable to us and our shareholders. | |
| ● | Your rights, liabilities and responsibilities as a shareholder will be governed by Israeli law and differ in some material respects from those under U.S. law. |
| 7 |
RISK FACTORS
An investment in our ordinary shares involves a high degree of risk. Prior to making a decision about investing in our ordinary shares, you should carefully consider the risks, uncertainties and assumptions set forth below. Additional risks and uncertainties not presently known to us, or that we currently see as immaterial, may also harm our business. If any of these risks occur, our business, financial condition and operating results could be harmed, the trading price of our ordinary shares could decline and you could lose part or all of your investment.
Risks Related to Our Financial Position and Capital Requirements
We are a clinical-stage biopharmaceutical company with a history of operating losses. We expect to incur significant additional losses in the future and may never be profitable.
We are a clinical-stage biopharmaceutical company with an operating history limited to pre-clinical and clinical drug development and no approved products. In addition, we have limited operating experience and have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the pharmaceutical industry. We have funded our research and development programs and operations to date primarily through proceeds from private placements and public offerings. We currently have no products approved for marketing in the United States or any other jurisdiction and have not generated any revenue from product sales to date, although we have generated revenue from our licensing agreement with Samil Pharm. Co., Ltd., or Samil. We have incurred operating losses in each year since the inception of our predecessor in 2000. Our loss attributable to holders of our ordinary shares for the years ended December 31, 2020, and 2021 was approximately $28.8 million and $32.5 million, respectively. As of December 31, 2021, we had an accumulated deficit of $168.2 million. Substantially all of our operating losses resulted from costs incurred in connection with our development program and from general and administrative costs associated with our operations.
Our ability to become profitable depends upon our ability to generate revenue in excess of our expenses. To date, we have not generated any revenue, excluding the licensing revenue we recorded in connection with that certain Samil Agreement (as defined below), as our lead product candidate, Aramchol is still in clinical development and has not been approved by the FDA, nor has any other product candidate. We do not know when, or if, we will generate any revenue from sales of Aramchol, Amilo-5MER and/or any other product candidate. We do not expect to generate revenue other than subsequent royalties and/or milestones that can be earned in connection with the Samil Agreement or other potential license agreements, unless and until we, or an ultimate third-party licensor or acquirer, obtain regulatory and marketing approval of, and commercialize, Aramchol, Amilo-5MER or any other product candidate. We will continue to incur significant research and development and general and administrative expenses related to our operations. We expect to continue to incur losses for the foreseeable future, which may be significant, and these losses will likely increase as we:
| ● | manage our ongoing ARMOR Study and any additional clinical trials for Aramchol and Amilo-5MER, or any other product candidate and initiate additional research and development programs; | |
| ● | seek regulatory approvals for Aramchol, Amilo-5MER or any other product candidate; | |
| ● | implement internal systems and infrastructures, including, without limitation, hiring of additional personnel as needed and developing sales and marketing functions if and when Aramchol or any other product candidate receives applicable regulatory approval and we opt to commercialize it ourselves; | |
| ● | seek to in-license additional products or technologies to develop; | |
| ● | hire additional management and other personnel; and | |
| ● | move towards commercialization of Aramchol or any other product candidate. |
| 8 |
We may out-license Aramchol, Amilo-5MER or any other product candidate including through a territorial license, a worldwide license, or a license for a particular indication, before it is approved by any applicable regulatory agency, commercialized and/or generates revenue, depending on a number of factors, including, but not limited to, our ability to:
| ● | demonstrate a compelling and/or novel, pre-clinical, unique mechanism of action of Aramchol, Amilo-5MER or any other product candidate; | |
| ● | obtain adequate clinical results from and progress from the clinical development of Aramchol or any other product candidate; | |
| ● | develop and obtain regulatory approvals in the countries and for the uses we intend to pursue for Aramchol, Amilo-5MER or any other product candidate; | |
| ● | contract for the manufacture of commercial quantities of Aramchol, Amilo-5MER or any other product candidate by a current good manufacturing practice, or cGMP, compliant manufacturing facility at acceptable cost levels if marketing approval is received; and | |
| ● | establish external, and potentially in the future, internal, sales and marketing capabilities to effectively market and sell Aramchol, Amilo-5MER or any other product candidate in the United States and other countries. |
Even if Aramchol, Amilo-5MER or any other product candidate is approved for commercial sale, it may not gain market acceptance or achieve commercial success. In addition, we anticipate incurring significant costs associated with seeking regulatory approval and commercialization. We may not achieve profitability soon after generating product revenue, if ever. If we are unable to generate product revenue, we will not become profitable and would be unable to continue operations without additional funding.
We expect our research and development expenses to significantly increase as we further progress in our ARMOR Study and initiation of any other pre-clinical or clinical trials. In addition, if we obtain marketing approval for Aramchol, Amilo-5MER or any other product candidate and opt to commercialize it ourselves, we will likely initially incur significant expenses associated with outsourcing sales, marketing and manufacturing functions to third parties, as well as continued research and development expenses. Furthermore, we expect to incur additional costs associated with operating as a public company. As a result, we expect to continue to incur significant and increasing operating losses for the foreseeable future. Because of the numerous risks and uncertainties associated with developing pharmaceutical products, we are unable to predict the extent of any future losses or when we will become profitable, if at all.
Our limited operating history makes it difficult to evaluate our business and prospects.
Our operating history is limited to pre-clinical and clinical development of two products, and our operations to date have been limited primarily to research and development, raising capital and recruiting scientific and management personnel and third-party partners. Therefore, it may be difficult to evaluate our business and prospects. We have not yet demonstrated an ability to commercialize or obtain regulatory approval for any product candidate. Consequently, any predictions about our future performance may not be accurate, and you may not be able to fully assess our ability to complete development and/or commercialize our product candidates, obtain regulatory approvals or achieve market acceptance or favorable pricing for our product candidates.
| 9 |
We have not yet commercialized any products and we may never be able to do so, and even if we do, the products may not gain market acceptance.
We have not yet commercialized any products and we may never be able to do so. We do not know when or if we will complete development of Aramchol, Amilo-5MER or any other product candidate, obtain regulatory approval, or successfully commercialize any approved products. Even if we are successful in developing products that are approved for marketing, we will not be successful unless these products gain market acceptance for appropriate indications at favorable reimbursement rates. The degree of market acceptance for these products will depend on a number of factors, including:
| ● | the timing and scope of regulatory approvals in the countries we intend to pursue with respect to the commercialization of Aramchol, Amilo-5MER or any other product candidate, including the indications for which they are approved; | |
| ● | the competitive environment; | |
| ● | the ability for Aramchol, Amilo-5MER or any other product candidate to be manufactured, whether by us or third parties, in compliance with applicable regulatory requirements, including cGMP; | |
| ● | our ability to effectively promote Aramchol or any other product candidate, whether directly or using third parties, consistent with the approved indications and labeling in the countries in which we intend to pursue approval; | |
| ● | the acceptance by the medical community of the safety and clinical efficacy of Aramchol, Amilo-5MER or any other product candidate and their potential advantages over other therapeutic products; | |
| ● | the development of a non-invasive method for diagnosing NASH as an alternative to the current gold standard of liver biopsy, which we view as a rate-limiting factor to complete market uptake because of its expense and its risks and discomfort to patients; | |
| ● | the adequacy and success of distribution, sales and marketing efforts, including through strategic agreements with pharmaceutical and biotechnology companies; and | |
| ● | the pricing and reimbursement policies of government and third-party payors, such as insurance companies, health maintenance organizations and other plan administrators. |
Physicians, patients, third-party payors or the medical community in general may be unwilling to accept, utilize or recommend, and in the case of third-party payors, reimburse any of our planned future products. As a result, we are unable to predict the extent of future losses or the time required to achieve profitability, if at all. Even if we successfully develop one or more products, we may not become profitable.
We will need substantial, additional capital in the future. If additional capital is not available, we will have to delay, reduce or cease operations.
As of December 31, 2021, we had a net working capital of $30.2 million, cash and cash equivalents of $2.9 million, restricted cash of $0.1 million, and marketable debt securities of $31.9 million. Based on our current operating plan, we currently estimate that our cash position will support our current clinical trials and operations as currently conducted for more than 12 months from the date of issuance of this annual report. We will need to raise substantial, additional capital to fund our operations and to develop Aramchol and Amilo-5MER for, and beyond their current development stage, and ultimately commercialize them, if we opt to do so ourselves. In addition, we are looking to expand our current research and development focus and clinical operations as well as the development of Aramchol or any other product candidate for other indications or development of other molecules and/or combination of Aramchol with other molecules for NASH or other liver and inflammatory diseases as well as non-invasive biomarkers, which we expect will also require additional capital. Our future capital requirements may be substantial and will depend on many factors including:
| ● | adhering to patient recruitment in our clinical trials; | |
| ● | our clinical trials results; |
| 10 |
| ● | the acceptance of any amendments to our Investigational New Drug application, or IND, or foreign equivalent for the ARMOR Study by the FDA and any other foreign regulatory authority and the acceptance of any other IND or foreign equivalent for any other product candidate; | |
| ● | developing Aramchol and combination of it for the treatment of other conditions or indications beyond NASH; | |
| ● | the cost of filing and prosecuting patent applications and the cost of defending our patents; | |
| ● | the cost of prosecuting infringement actions against third parties; | |
| ● | the cost, timing and outcomes of seeking marketing approval of Aramchol or any other product candidate; | |
| ● | the costs associated with commercializing Aramchol or any other product candidate if we receive marketing approval, and choose to commercialize our product candidates ourselves, including the cost and timing of establishing external, and potentially in the future, internal, sales and marketing capabilities to market and sell our product candidates; | |
| ● | the costs associated with any product liability or other lawsuits related to Aramchol or any other product candidate; | |
| ● | the costs associated with post-market compliance with regulatory requirements, and of addressing any allegations of non-compliance by regulatory authorities in countries where we plan to market and sell Aramchol or any other product candidate; | |
| ● | the demand for Aramchol, Amilo-5MER or any other product candidate; | |
| ● | the costs associated with developing, in-licensing, or acquiring other research and development programs; | |
| ● | the expenses needed to attract and retain skilled personnel; | |
| ● | the costs associated with being a public company; and | |
| ● | the impact of the COVID-19 pandemic and the Russian invasion of Ukraine, which may exacerbate the magnitude of the factors discussed above. |
Under General Instruction I.B.5 to Form F-3, or the Baby Shelf Rule, the amount of funds we can raise through primary public offerings of securities in any 12-month period using our registration statement on Form F-3 is limited to one-third of the aggregate market value of the ordinary shares held by non-affiliates of the Company. As of April 27, 2022, our public float was approximately $38.2 million, based on 21,591,375 ordinary shares held by non-affiliates and a price of $1.77 per share, which was the last reported sale price of our ordinary shares on the Nasdaq Capital Market on April 5, 2022. We therefore are limited by the Baby Shelf Rule as of the filing of this annual report, until such time as our public float exceeds $75 million. If we are required to file a new registration statement on another form, we may incur additional costs and be subject to delays due to review by the SEC Staff.
| 11 |
Changing circumstances may cause us to consume capital significantly faster than we currently anticipate, such as losing our Small and Medium Enterprise status at the EMA, which entitles us to significant fee reductions. Because there are numerous risks and uncertainties associated with the development and commercialization of Aramchol, Amilo-5MER or any other product candidate, we are unable to estimate the amount of increased capital outlays and operating expenditures associated with our anticipated clinical trials. We have no committed external sources of funds. Additional financing may not be available when we need it or may not be available on terms that are favorable to us and additional financing may cause significant dilution to our existing shareholders. If adequate funds are not available to us on a timely basis, or at all, we may be required to terminate or delay planned or ongoing clinical trials or other development activities for Aramchol, Amilo-5MER or any other product candidate.
Raising additional capital may be costly or difficult to obtain and will dilute current shareholders’ ownership interests, potentially substantially.
Any debt, equity or structured financing that we may need or desire may not be available on terms favorable to us, or at all. If we obtain funding through a strategic collaboration or licensing arrangement, we may be required to relinquish our rights to certain of our technologies, products or marketing territories. If we are unable to obtain required additional capital, we may have to curtail our growth plans or cut back on existing business, and we may not be able to continue operating if we do not generate sufficient revenues from operations needed to stay in business.
We may incur substantial costs in pursuing future capital financing, including investment banking fees, legal fees, accounting fees, securities law compliance fees, printing and distribution expenses and other costs. We may also be required to recognize non-cash expenses in connection with certain securities we issue, such as convertible notes and warrants, which may adversely impact our capital structure, financial condition and results of operations.
Any additional capital raised through the sale of equity or equity-linked securities will dilute our current shareholders’ ownership in us, potentially substantially, and could also result in a decrease in the market price of our ordinary shares. The terms and conditions of those securities issued by us in future capital transactions may be more favorable to new investors and may include the issuance of warrants or other derivative securities, which may have a further dilutive effect.
We are unable to estimate our long-term capital requirements due to uncertainties associated with the development and commercialization of Aramchol, Amilo-5MER or any other product candidate. If we fail to obtain necessary funds for our operations, we will be unable to develop and commercialize Aramchol, Amilo-5MER or any other product candidate.
Our long-term capital requirements are expected to depend on many potential factors, including, among others:
| ● | the number of product candidates in development; | |
| ● | the size, duration and scope of existing and future clinical trials and pre-clinical studies; | |
| ● | the regulatory pathway for the approval of Aramchol, Amilo-5MER or any other product candidate; | |
| ● | the results of our clinical trials, which are unpredictable in product candidate development; | |
| ● | our ability to successfully commercialize Aramchol, Amilo-5MER or any other product candidate, including securing commercialization and out-licensing agreements with third parties and favorable pricing and market share; | |
| ● | the progress, success and cost of our clinical trials and research and development programs, including those associated with milestones and royalties; |
| 12 |
| ● | the costs, timing and outcome of regulatory review and obtaining regulatory approval of Aramchol, Amilo-5MER or any other product candidate and addressing regulatory and other issues that may arise post-approval; | |
| ● | the breadth of the labeling, assuming that Aramchol, Amilo-5MER or any other product candidate are approved for commercialization by a relevant regulatory authority, which may not occur; | |
| ● | our need, or decision, to acquire or in-license complementary technologies or new platform technologies or product candidates; | |
| ● | the costs of enforcing our issued patents and defending intellectual property-related claims; | |
| ● | the costs of investigating patents that might block us from developing potential product candidates; | |
| ● | the costs of recruiting and retaining qualified personnel; | |
| ● | the costs associated with contracting with third parties to manufacture the product and to perform other necessary services; | |
| ● | our revenue, if any; and | |
| ● | our consumption of available resources more rapidly than currently anticipated, resulting in the need for additional funding sooner than anticipated. |
If we are unable to obtain the funds necessary for our operations, we will be unable to develop and commercialize Aramchol, Amilo-5MER or any other product candidate which would materially and adversely affect our business, liquidity and results of operations.
Risks Related to Our Business, Industry and Regulatory Requirements
The clinical trial process is complex and expensive, and commencement and completion of clinical trials can be delayed or prevented for a number of reasons.
We may not be able to complete or commence the clinical trials that would support our submission of an NDA to the FDA, a Marketing Authorization Application or MAA, to the EMA or MHRA or any similar submission to regulatory authorities in other countries. Drug development is a long, expensive and uncertain process, and delay or failure can occur at any stage of any of our clinical trials. The fact that the FDA, EMA, MHRA or other regulatory authorities permit a company to conduct human clinical trials is no assurance or guarantee that the trials will be successful. On the contrary, most candidate drugs that begin clinical trials do not prove to be successful and do not result in the filing of an NDA, MAA or similar filing. Drug candidates that successfully complete one phase of clinical trials may prove unsuccessful at a subsequent phase. Human clinical trials are very expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements and in part because the results of clinical trials are inherently uncertain and unpredictable. In addition, the design of a clinical trial can determine whether its results will support approval of a product, and flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced. Regulatory authorities, such as the FDA, may decline to permit a clinical trial to proceed or may suspend a clinical trial that it has previously permitted to proceed. Additionally, the clinical trial process is time-consuming, and failure can occur at any stage of the trials. We may encounter problems that cause us to abandon or repeat clinical trials. The commencement and completion of clinical trials may be delayed by several factors, including:
| ● | difficulties obtaining regulatory authorization to commence a clinical trial or complying with regulatory requirements for clinical trials or with the conditions imposed by a regulatory authority regarding the scope, duration or conduct of a clinical trial; |
| 13 |
| ● | delays in reaching or failing to reach agreement on acceptable terms with prospective contract research organizations, or CROs, and trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; | |
| ● | insufficient or inadequate supply or quality of a product candidate or other materials necessary to conduct our clinical trials; | |
| ● | difficulties in obtaining institutional review board, or IRB, approval to conduct a clinical trial at a prospective site; | |
| ● | challenges in recruiting and enrolling patients to participate in clinical trials for a variety of reasons, including size and nature of patient population, proximity of patients to clinical sites, eligibility and exclusion criteria for the trial, nature of trial protocol, the availability of approved effective treatments for the relevant disease and competition from other clinical trial programs for similar indications; | |
| ● | challenges in identifying or recruiting sufficient study sites or investigators for clinical trials; and | |
| ● | lack of adequate funding to continue our clinical trials. |
Even though we initiated the Phase 3 ARMOR Study, the ARMOR Study may still be terminated as a result of, but not limited to, safety signals, lack of efficacy, uncertainties with regard to the regulatory pathway for the approval of NASH drugs, or commercial considerations based on the cost-benefit of continuing to run the study. In addition, the ARMOR Study or other clinical trials may be suspended or terminated by us, the FDA or other regulatory authorities, the principal investigator at a site, the IRBs at the sites where such boards are overseeing a trial or the data safety monitoring board, or the DSMB, that is overseeing the clinical trial at issue, or other regulatory authorities due to a number of factors, including:
| ● | irregularities in conducting a clinical trial, including by way of example, failure to conduct the clinical trial in accordance with regulatory requirements, in particular good clinical practice requirements, or GCP, or the FDA-authorized clinical protocols; | |
| ● | negative findings upon inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities; | |
| ● | safety issues or lack of clinical drug activity or effectiveness; and | |
| ● | lack of adequate funding to continue the clinical trials. |
To date, we have already experienced material delays in both the ARMOR Study largely related to significantly slower than expected recruitment and the ARREST Study largely related to significantly slower than expected recruitment and the length of time required to obtain regulatory authorizations to proceed with clinical trials. We may experience further delays in any or all of our clinical trials and there can be no assurance that we will not experience such risks in the future as we progress with our planned clinical trials.
| 14 |
Furthermore, positive results in previous clinical studies of our product candidates may not be predictive of similar results in future clinical trials. Also, interim results, if at all, during a clinical trial do not necessarily predict final results. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials even after achieving promising results in early- and mid-stage development. Accordingly, the results from the completed pre-clinical studies and clinical trials for our product candidates may not be predictive of the results we may obtain in later stage trials. Our clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical and/or pre-clinical trials, or to even terminate the development program entirely. Many companies that believed their product candidates performed satisfactorily in pre-clinical and clinical studies have nonetheless failed to obtain FDA or EMA, or other regulatory agency, approval for their products.
In addition, we or regulatory authorities may suspend our clinical trials at any time if it appears that we are exposing participants to unacceptable health risks or if the regulatory authorities find deficiencies in our regulatory submissions or the conduct of such trials. Any suspension of clinical trials will delay possible regulatory approval, if any, and adversely impact our ability to develop products and generate revenue.
We are developing Aramchol for the treatment of NASH, an indication for which there are no approved products, and there is significant uncertainty regarding the regulatory approval process. This makes it difficult to predict the timing and costs of the clinical development of Aramchol for the treatment of NASH.
Pharmaceutical products generally are subject to rigorous nonclinical testing and clinical studies and other approval procedures mandated by the FDA and foreign regulatory authorities. We are developing Aramchol for the treatment of NASH, an indication for which there are no approved products.
In September 2019, we initiated the ARMOR Study. As part of our ongoing review process, we have been in an ongoing dialogue with the FDA with regard to the ARMOR Study trial design, statistical analysis plan and the conduct of the trial. We have made certain amendments to the ARMOR Study including the addition of an open label part and an extension of the histology-based phase to 72 weeks while reducing the number of patients in the histology-based phase from 1200 to 1000. As a result of the changes to the ARMOR Study design, this has resulted in extending the duration of the ARMOR Study and has made the clinical trial process more expensive. In addition, we plan to transition from Aramchol free acid to Aramchol meglumine (salt), and this may further result in additional delays in the completion of the ARMOR Study and may result in further clinical trial expenses. Our anticipated development costs would likely increase if development of Aramchol or any future product candidate is delayed because we are required by the FDA or any other regulatory agency to perform studies or trials in addition to, or different from, those that we currently anticipate.
Although there are guidelines issued by the FDA for the development of drugs for the treatment of NASH, it is our current assessment that despite considerable efforts from the scientific community and regulatory agencies, there are significant uncertainties that remain unresolved with regards to the conduct of NASH Phase 3 registrational studies. These risks include dependence on biopsies as the primary surrogate endpoint which are subjective in nature and prone to sampling errors and inadequacy; high screen failure rates which pose a burden to patients, investigators, and study budget; lack of validated biomarkers and the heterogenous population included under the NASH diagnosis. Moreover, pathology plays a critical role in NASH clinical trials with histology being the current reference method to determine inclusion in trials and change in disease activity and fibrosis stage. Manual histological review is complex, subjective, and prone to inter- and intra-reader variability and error. Existing pathology scoring systems and practices show only moderate to fair reproducibility, limiting their utility for clinical research and practice.
In addition, the FDA has indicated that the results of the ARMOR Study must be unequivocal and highly persuasive for a single Phase 3 study to support approval of an NDA. Therefore, even if the ARMOR Study meets all of its statistical goals and protocol endpoints, the FDA may not view the results as sufficient to support an NDA.
We expect that the path for regulatory approval for NASH drugs may continue to evolve in the near term as we and other companies in late-stage development of NASH drugs refine our regulatory approval strategies and interact with the FDA and other regulatory authorities. In particular, FDA expectations about interpretation of liver biopsy data may evolve especially as more information is published about the inherent variability in interpretation of liver biopsy data. Certain of our competitors have experienced regulatory setbacks for NASH therapies following communications from the FDA. See also “Item 4. Information on the Company—Competition.” Even after we receive and incorporate guidance from the FDA or other regulatory authorities, they could disagree that we have satisfied their requirements, which may require us to complete additional preclinical studies or clinical trials or impose stricter approval conditions than we currently expect. Furthermore, as the path for regulatory approval for NASH drugs evolves, it may impact our ARMOR Study in ways that could significantly increase the development costs and call into question the future viability of continuing to conduct the ARMOR Study. Additionally, we are considering more robust changes to our development program for NASH. Changes may include focusing on higher risk patients (F3), evaluating patients with compensated cirrhosis (F4) as well as changes to study design such as two smaller studies instead of one pivotal study and the addition of a combination arm.
| 15 |
Any additional delays in the completion of the ARMOR Study or any additional preclinical studies or clinical trials would require us to expend substantial additional resources and could significantly extend the timeline for clinical development prior to market approval. As a result of the foregoing, the research and development, preclinical studies and clinical testing of Aramchol and any other product candidate is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the development process. If we experience delays in the completion of, or if we terminate, any of our clinical trials, this would have a material adverse effect on our business, liquidity, operating results and financial condition and may force us to cease operations.
We depend largely on the success of our lead product candidate, Aramchol, and we may not obtain regulatory approval of Aramchol.
We have invested almost all of our efforts and financial resources in the research and development (clinical and pre-clinical) of our lead product candidate, Aramchol. As a result, our business is largely dependent on the success of the ARMOR Study and our ability to complete the development of, obtain regulatory approval for and successfully commercialize Aramchol in a timely manner. The process to develop, obtain regulatory approval for and commercialize Aramchol is long, complex, costly and uncertain as to its outcome.
The research, development, testing, clinical trials, manufacturing, labeling, approval, sale, marketing and distribution of drugs are subject to extensive regulation by the FDA and other regulatory agencies in other countries. These regulations differ from jurisdiction to jurisdiction. We have not received marketing approval for Aramchol in any jurisdiction. We are not permitted to market Aramchol, or any other product candidate, in the United States until we receive approval of a New Drug Application, or NDA, from the FDA, or in any foreign countries until we receive the requisite approval from the respective regulatory agencies in such countries. The results of clinical trials may be unsatisfactory, and even if we believe those clinical trials to be successful, the FDA, or other regulatory authorities, may not grant marketing authorization should we be in a position to request it.
The requirements and length of time for approval vary in different jurisdictions and could involve additional studies of Aramchol beyond those we currently anticipate, including potentially post-approval studies. The time required to obtain approval in other countries might differ from that required to obtain FDA approval in the United States. The marketing approval process in other countries may include all of the risks detailed above regarding FDA approval as well as other risks. In particular, in many countries outside the United States, it is required that a product receive pricing and reimbursement approval before the product can be commercialized. This can result in substantial delays in such countries. In other countries, product approval depends on showing superiority to an approved therapy. This can result in significant expense to conduct complex clinical trials. Finally, we do not have any products approved for sale in any jurisdiction, including international markets, and we do not have experience in obtaining regulatory approval in international markets. If we fail to comply with regulatory requirements in international markets or to obtain and maintain required approvals or if regulatory approvals in international markets are delayed, our target market will be reduced and our ability to realize the full market potential of Aramchol or any other product candidate will be harmed.
Marketing approval in one jurisdiction does not ensure marketing approval in another, but a failure or delay in obtaining marketing approval in one jurisdiction may have a negative effect on the regulatory process in others. Failure to obtain marketing approval in other countries or any delay or setback in obtaining such approval would impair our ability to develop foreign markets for Aramchol. This would reduce our target market and limit the full commercial potential of Aramchol.
| 16 |
Commencement of our ARMOR Study in jurisdictions outside the United States is subject to acceptance of the foreign equivalent of our IND by regulatory authorities.
In September 2019, we initiated the ARMOR Study and in late 2020 we added an open label part to the study. In the event that the FDA or any other regulatory authority requires us to complete additional preclinical and/or clinical studies or we are required to satisfy other FDA or other regulatory requests, the start of the ARMOR Study in the applicable jurisdiction or any of our other programs may be delayed or not started at all. For example, certain regulatory agencies in Europe have required that we conduct additional clinical studies prior to initiating ARMOR in those jurisdictions. Even after we receive and incorporate guidance from these regulatory authorities, the FDA or other regulatory authorities could disagree that we have satisfied their requirements to commence our clinical trial or change their position on the acceptability of our planned trial design or the clinical endpoints selected, which may require us to complete additional preclinical studies or clinical trials or impose stricter approval conditions than we currently expect. As a result of the foregoing, the research and development, preclinical studies and clinical testing of any product candidate is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the development process.
We may be forced to abandon development of Aramchol or any other product candidate which would have a material adverse effect on our business and may force us to cease operations.
Upon the completion of any clinical or pre-clinical trial and/or tests, the results might not support the desired indications for use. Further, success in earlier clinical trials does not ensure that later clinical trials will be successful, and the results of later clinical trials may not replicate the results of prior clinical trials or pre-clinical testing. In addition, the design of a clinical trial can determine whether its results will support approval of a product, and flaws in the design of a clinical trial or variability in interpretation of results may not become apparent until the clinical trial is well advanced. The clinical trial process may fail to demonstrate that Aramchol or any other product candidate is sufficiently safe and/or effective for the indications we seek to receive FDA or other regulatory approval. Any such failure may cause us to abandon Aramchol or any other product candidate and may delay development of other potential product candidates. Any delay in, or termination or suspension of, our clinical trials may delay the requisite filings with the FDA or other regulatory agencies and, ultimately, our ability to commercialize Aramchol or any other product candidate and generate product revenues. In September 2019, we initiated the ARMOR Study and in late 2020 added an open label part. If the results of the ARMOR Study or any other study, including in any interim readout, are not sufficiently compelling, then the completion of development of our product candidates may be significantly delayed or abandoned which would have material adverse effect on our business, liquidity, operating results and financial condition and may force us to cease operations.
We have in the past and may in the future develop Aramchol in combination with other therapies, which exposes us to additional risks.
We have in the past and may in the future develop Aramchol in combination with investigational therapies. We will not be able to market and sell Aramchol or any product candidate we develop in combination with an unapproved therapy for a combination indication if that unapproved therapy does not ultimately obtain marketing approval either alone or in combination with our product candidate. In addition, unapproved therapies face the same risks described with respect to Aramchol or any other product candidate currently in development and clinical trials, including the potential for serious adverse effects, delay in their clinical trials and lack of FDA, EMA or MHRA approval. If the FDA, EMA, MHRA or comparable foreign regulatory authorities do not approve these other drugs or revoke their approval of, or if safety, efficacy, quality, manufacturing or supply issues arise with the product candidates we choose to evaluate in combination with our product candidate we develop, we may be unable to obtain approval of or market such combination therapy.
| 17 |
The lack of a reliable non-invasive method for the diagnosis of NASH and fibrosis is likely to present a major challenge to Aramchol’s market penetration, if ever commercialized.
Liver biopsy is the standard approach for the diagnosis of inflammation and fibrosis associated with NASH. However, the procedure-related morbidity and, in rare cases, mortality, sample errors, costs, patient discomfort and thus lack of patient interest in undergoing the procedure limit its use. As such, only patients with a high risk of NASH, which includes patients with metabolic syndrome and an indication of Non-Alcoholic Fatty Liver Disease, or NAFLD, are generally sent for liver biopsy. Because NASH tends to be asymptomatic until the disease progresses, many individuals with NASH remain undiagnosed until the disease has reached its late stages, if at all. The lack of a reliable non-invasive method for the diagnosis of NASH and fibrosis is likely to present a major challenge to Aramchol’s market penetration, as many practitioners and patients may not be aware that a patient suffers from NASH and requires treatment. As such, use of Aramchol might not be as wide-spread as our actual target market and this may limit the commercial potential of Aramchol.
A further challenge to Aramchol’s market penetration is that currently a liver biopsy is the standard approach for measuring improvement in NASH patients. Because it would be impractical to subject all patients that take Aramchol, when and if it approved, to regular and repeated liver biopsies, it will be difficult to demonstrate Aramchol’s effectiveness to practitioners and patients unless and until a reliable non-invasive method for the diagnosis and monitoring of NASH becomes available, as to which there can be no assurance.
While we, and other companies in the industry are currently working on advancing non-invasive diagnostic approaches, none of these has been clinically validated, and the timetable for commercial validation, if at all, is uncertain. Moreover, such diagnostics may also be subject to regulation by FDA or other regulatory authorities as medical devices and may require premarket clearance or approval. See also “Item 3. Key Information—Risk Factors—Risk Factors—Risks Related to Our Business, Industry and Regulatory Requirements —- We are developing Aramchol for the treatment of NASH, an indication for which there are no approved products, and there is significant uncertainty regarding the regulatory approval process. This makes it difficult to predict the timing and costs of the clinical development of Aramchol for the treatment of NASH.”
Our Amilo-5MER program is being conducted under a license agreement with Yissum Research Development Company of the Hebrew University of Jerusalem, or Yissum.
Our Amilo-5MER program is being conducted under a license agreement from Yissum and is subject to various additional obligations, including obligations with respect to funding, development and commercialization activities, and payment obligations upon entering into the license agreement and achievement of certain milestones and royalties on product sales. Furthermore, if the license agreement is terminated or breached, we may:
| ● | lose our rights to research, develop or commercialize Amilo-5MER; | |
| ● | not be able to secure trade secret protection for Amilo-5MER; | |
| ● | experience significant delays in the development or commercialization of Amilo-5MER; or may have to cease development entirely; | |
| ● | incur liability for damages. |
Additionally, even if not terminated or breached, our intellectual property licenses may be subject to disagreements over contract interpretation which could narrow the scope of our rights to the relevant intellectual property or technology or increase our financial or other obligations. If we experience any of the foregoing, it could have a materially adverse effect on our business.
| 18 |
In January 2022, we announced the completion of our Phase 1 clinical trial of Amilo-5MER. The Phase 1 study was conducted in a single-center, using a double-blind, randomized, placebo-controlled design. Results demonstrated that all doses of Amilo-5MER were well tolerated with no clinically significant adverse events and none considered related to the investigational product. Due to its early stage of development, Amilo-5MER will require significant additional research, development, manufacturing, preclinical and clinical testing, marketing authorization, and commitment of significant additional resources prior to any commercialization. These activities will require significant cash for which we will need to raise additional capital. In addition, Amilo-5MER is prone to the risks of failure inherent in pharmaceutical product development, including the possibility that Amilo-5MER will not be shown to be sufficiently safe and effective for approval by regulatory authorities.
If we acquire or in-license additional technologies or product candidates, we may incur significant, incremental expenses, may have integration difficulties and may experience other risks that could harm our business and results of operations.
We are currently evaluating the acquisition or in-licensing of additional product candidates and technologies. Any product candidate or technologies we in-license or acquire will likely require additional development efforts prior to commercial sale, including extensive pre-clinical or clinical testing, or both, and approval by the FDA and applicable foreign regulatory authorities, if any. All product candidates are prone to risks of failure inherent in pharmaceutical product development, including the possibility that the product candidate, or product developed based on in-licensed technology, will not be shown to be sufficiently safe and effective for approval by regulatory authorities. In addition, we cannot assure that any product candidate that we develop based on acquired or in-licensed technology that is granted regulatory approval will be manufactured or produced economically, successfully commercialized or widely accepted or competitive in the marketplace. Moreover, integrating any newly acquired or in-licensed product candidates could be expensive and time-consuming. If we cannot effectively manage these aspects of our business strategy, our business may not succeed.
Obtaining approval of an NDA, or other regulatory approval, even after clinical trials that are believed to be successful, is an uncertain process.
Even if we complete our planned clinical trials and believe that the clinical data confirms that Aramchol, Amilo-5MER or any other product candidate is sufficiently safe and effective for its intended use or uses, obtaining approval of an NDA, or other regulatory approval, is an extensive, lengthy, expensive and uncertain process, and the FDA and other regulatory agencies may delay, limit or deny approval of such product candidate for many reasons, including, without limitation, the fact that:
| ● | we may not be able to demonstrate to the satisfaction of the applicable regulatory agencies that the product candidate is safe and effective for treatment of the targeted indication in patients; | |
| ● | the results of clinical trials may not meet the level of statistical significance or clinical significance required by the applicable regulatory agencies for approval; | |
| ● | the applicable regulatory agencies may disagree with the number, design, size, conduct or implementation of our clinical trials; | |
| ● | the applicable regulatory agencies may not find the data from pre-clinical studies and clinical trials sufficient to demonstrate the clinical and other benefits outweigh its safety risks; | |
| ● | the applicable regulatory agencies may disagree with our interpretation of data from pre-clinical studies or clinical trials; | |
| ● | the applicable regulatory agencies may not accept data generated at our clinical trial sites; |
| 19 |
| ● | the data collected from pre-clinical studies and clinical trials may not be sufficient to support the submission of an NDA or similar regulatory application; | |
| ● | the applicable regulatory agencies may not schedule an advisory committee meeting in a timely manner or the advisory committee may recommend against approval of our application or may recommend that the applicable regulatory agencies require, as a condition of approval, additional pre-clinical studies or clinical trials, limitations on approved labeling or distribution and use restrictions; | |
| ● | the applicable regulatory agencies may require development of a risk evaluation and mitigation strategy, or REMS, as a condition of approval; | |
| ● | the applicable regulatory agencies may require simultaneous approval for both adults and children, which would delay required approvals, or we may have successful clinical trial results for adults, but not children, or vice versa; | |
| ● | the applicable regulatory agencies may change their approval policies or adopt new regulations that may impede consideration or approval of our NDA, or similar regulatory application; | |
| ● | the applicable regulatory agencies may identify deficiencies in the manufacturing processes or facilities of third-party manufacturers, or suppliers of active pharmaceutical ingredients, or APIs, with which we enter into agreements for clinical and commercial supplies; and | |
| ● | the applicable regulatory agencies may require post-marketing approval studies, such as Phase 4 clinical trials, in connection with Aramchol or any other product candidate. |
Before we can submit an NDA to the FDA or a similar approval application to other regulatory authorities, as applicable, we (or our commercialization partner, as the case may be) must conduct one or more clinical trials that will be substantially broader than our prior completed trials. We will also need to agree on a protocol with the FDA or any other regulatory authorities for any clinical trial(s) before commencing any such trial. Clinical trials frequently produce unsatisfactory results even though prior clinical trials were successful. Therefore, the results of any prior trial or any future clinical trials that we may conduct may or may not be successful. The applicable regulatory agencies may suspend all clinical trials or require that we conduct additional clinical, pre-clinical, manufacturing, validation or drug product quality studies and submit data from these additional studies before considering or reconsidering the NDA or similar regulatory application. Depending on the extent of these, or any other studies, approval of any applications that we submit may be delayed by several years, or may require us to expend more resources than we have available. It is also possible that additional studies, if performed and completed, may not be considered sufficient by the applicable regulatory agencies to provide regulatory approval. If any of these outcomes occur, we would not receive approval for Aramchol, Amilo-5MER or any other product candidate and may be forced to cease operations.
Even if we obtain regulatory approval for Aramchol, Amilo-5MER or any other product candidate, the approval might contain significant limitations related to the indications for use for which the drug is approved, use restrictions including, without limitation, for certain labeled populations, age groups, warnings, precautions or contraindications, or may be subject to significant post-marketing studies or risk mitigation requirements. If we are unable to successfully commercialize Aramchol, Amilo-5MER or any other product candidate, we may be forced to cease operations.
| 20 |
Our product candidates may produce undesirable side effects or have other properties that could delay or prevent its regulatory approval or result in significant negative consequences following marketing approval, if any, which could substantially increase commercialization costs or even force us to cease operations.
Undesirable side effects caused by Aramchol or any other product candidate could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or applicable foreign regulatory authorities. To date, we have completed seven clinical trials of Aramchol, and additionally one proof of concept study in patient with gallstones, and a Phase 2a, investigator initiated clinical trial were completed. Although we have not seen any evidence of reactions causing a safety concern in our completed clinical trials, it is possible that the FDA may ask for additional data regarding any adverse events seen in our trials. Results of our future trials could reveal a high and unacceptable severity and prevalence of these or other side effects. In such an event, our trials could be suspended or terminated and the FDA or applicable foreign regulatory authorities could order us to cease further development of or deny approval for Aramchol, Amilo-5MER or any other product candidate for any or all targeted indications. The drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete future trials or result in potential product liability claims. Any of these occurrences may harm our business, financial condition and prospects significantly.
Even if Aramchol or any other product candidate receives marketing approval, we or others may later identify undesirable side effects caused by the product. In such an event, regulatory authorities may:
| ● | suspend or withdraw their approval of the product; | |
| ● | require the addition of labeling statements, such as warnings, so-called “black box warnings,” contraindications or restrictions on the product’s intended use; | |
| ● | require us to issue specific communications to healthcare professionals, such as “Dear Doctor” letters; | |
| ● | issue negative publicity regarding the affected product, including safety communications; | |
| ● | impose a risk evaluation and mitigation strategy (REMS), in the case of FDA, or similar risk management strategies in the case of foreign regulators; |
In addition to these potentially significant negative consequences, we could be required to change the way the product is administered, conduct additional pre-clinical studies or clinical trials or restrict or cease the distribution or use of the product, and/or be sued and held liable for harm caused to patients. The foregoing or other events could prevent us from achieving or maintaining market acceptance of the affected product candidate and could substantially increase commercialization costs or even force us to cease operations.
If we encounter difficulties enrolling patients in our clinical trials, our clinical development activities could be delayed or otherwise adversely affected.
Patient enrollment, a significant factor in the timing of clinical trials, is affected by many factors including the size and nature of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, patient willingness to undergo a liver biopsy in our NASH trials, competing clinical trials and clinicians’ and patients’ perceptions as to the potential advantages and disadvantages of the product candidate being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating, and actual or threatened public health emergencies and outbreaks of disease (including, for example, the COVID-19 pandemic). Potential patients for Aramchol, Amilo-5MER or any other product candidate may not be adequately diagnosed or identified with the diseases which we are targeting or may not meet the entry criteria for our studies.
We will be required to identify and enroll a sufficient number of patients in the U.S. with NASH for each of our planned clinical trials of Aramchol in this indication. We also may encounter difficulties in identifying and enrolling U.S. NASH patients who meet the eligibility criteria for our planned clinical trials. We may not be able to initiate or continue clinical trials if we are unable to locate a sufficient number of eligible patients to participate in the clinical trials required by the FDA or other foreign regulatory agencies. In addition, the process of finding and diagnosing patients may prove costly. To date, we have already experienced significant delays in our clinical trials largely related to significantly slower than expected recruitment and the length of time required to obtain regulatory authorizations to proceed with clinical trials. Our inability to enroll a sufficient number of patients for any of our clinical trials could result in further significant delays, additional expenses, or may require us to abandon one or more clinical trials.
| 21 |
Changes in regulatory requirements and guidance or unanticipated events during our clinical trials may occur, which may result in necessary changes to clinical trial protocols, which could result in increased costs to us, delay our development timeline or reduce the likelihood of successful completion of our clinical trials.
Changes in regulatory requirements or guidance or unanticipated events during our clinical trials may result in the need for us to amend clinical trial protocols. In December 2020, we announced the addition of an open label part to our ARMOR Study that involved a significant amendment to the clinical trial protocol. Amendments may require review and approval by regulators and/or IRBs, and re-consent subjects, which may adversely affect the cost, timing or successful completion of a clinical trial. If we experience delays in the completion of, or if we terminate, any of our clinical trials, this would have a material adverse effect on our business, liquidity, operating results and financial condition and may force us to cease operations. See also “Item 3. Key Information—Risk Factors—Risk Factors—Risks Related to Our Business, Industry and Regulatory Requirements - We are developing Aramchol for the treatment of NASH, an indication for which there are no approved products, and there is significant uncertainty regarding the regulatory approval process. This makes it difficult to predict the timing and costs of the clinical development of Aramchol for the treatment of NASH.”
Even if Aramchol, Amilo-5MER or any other product candidate that we develop, receives marketing approval, we will continue to face extensive regulatory oversight and requirements, and any such product may still face future regulatory risks or new requirements.
Even if we receive regulatory approval to market a particular product candidate, any such product will remain subject to extensive regulatory requirements, including requirements relating to manufacturing, labeling, packaging, adverse event reporting, storage, advertising, promotion, distribution and recordkeeping. Even if regulatory approval of a product is granted, the approval may be subject to limitations on the uses for which the product may be marketed or the conditions of approval, or may contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product, which could negatively affect us by reducing revenues or increasing expenses, and cause the approved product candidate not to be commercially viable. In addition, as clinical experience with a drug expands after approval, typically because it is used by a greater number and more diverse group of patients after approval than during clinical trials, side effects and other problems may be observed over time after approval that were not seen or anticipated during pre-approval studies. Any adverse effects observed after the approval and marketing of a product candidate could result in limitations on the use of the approved product, withdrawal of FDA approval of the previously approved product, or voluntary withdrawal from the marketplace of the approved product. Absence of long-term safety data may also limit the approved uses of Aramchol or any other product candidate, if any. If we fail to comply with the regulatory requirements of the FDA, and other applicable U.S. and foreign regulatory authorities, or previously unknown problems with any approved commercial products, manufacturers or manufacturing processes are discovered, we could be subject to administrative or judicially imposed sanctions or other setbacks, including the following:
| ● | suspension or imposition of restrictions on operations, including costly new manufacturing requirements; | |
| ● | refusal to approve pending applications or supplements to applications; | |
| ● | suspension of any ongoing clinical trials; |
| 22 |
| ● | suspension or withdrawal of marketing approval; | |
| ● | an injunction or imposition of civil or criminal penalties or monetary fines; | |
| ● | seizure or detainment of products; | |
| ● | banning or restriction of imports and exports; | |
| ● | issuance of warning letters or untitled letters; | |
| ● | suspension or imposition of restrictions on operations, including costly new manufacturing requirements; or | |
| ● | refusal to approve pending applications or supplements to applications. |
In addition, various aspects of our operations are subject to federal, state or local laws, rules and regulations, any of which may change from time to time. Costs arising out of any regulatory developments could be time-consuming and expensive and could divert management resources and attention and, consequently, could adversely affect our business operations and financial performance.
Delays in regulatory approval, limitations in regulatory approval and withdrawals of regulatory approval may have a material adverse effect on the Company. If we experience significant delays in testing or receiving approvals or sign-offs to conduct clinical trials, Aramchol or any other product candidate development costs will increase and our ability to out-license our product candidates may be impeded.
If we obtain approval to commercialize any product candidate outside of the United States or out-license a product candidate to additional territories outside the United States, a variety of risks associated with international operations could materially adversely affect our business.
If any product candidate is approved for commercialization outside the United States or we out-license a product candidate to additional territories outside the United States, we will likely enter into agreements with third parties to commercialize a product candidate outside the United States. We expect that we will be subject to additional risks related to entering into or maintaining international business relationships, including, without limitation:
| ● | different regulatory requirements for drug approvals in foreign countries; | |
| ● | differing U.S. and foreign drug import and export rules; | |
| ● | reduced protection for intellectual property rights in foreign countries; | |
| ● | unexpected changes in tariffs, trade barriers and regulatory requirements; | |
| ● | different reimbursement systems; | |
| ● | economic weakness, including inflation, or political instability in particular foreign economies and markets; | |
| ● | compliance with tax, employment, immigration and labor laws for employees living or traveling abroad; | |
| ● | foreign currency fluctuations, which could result in increased operating expenses and reduced revenues, and other obligations incident to doing business in another country; |
| 23 |
| ● | workforce uncertainty in countries where labor unrest is more common than in the United States; | |
| ● | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; | |
| ● | potential liability resulting from development work conducted by these distributors; | |
| ● | business interruptions resulting from geopolitical actions, including war and terrorism, or natural disasters, emergence of a pandemic, or other widespread health emergencies (or concerns over the possibility of such an emergency, including for example, the COVID-19 pandemic); and | |
| ● | risks associated with clinical co-development agreements in other jurisdictions prior to or post-regulatory approval. |
A failure to timely and effectively address the additional risks related to entering into or maintaining international business relationships could have a material adverse effect on our business, liquidity, operating results and financial condition.
If we receive marketing approval for a product candidate, sales will be limited unless the product achieves broad market acceptance.
The commercial success of a product candidate for which we obtain marketing approval from the FDA, or other regulatory authorities, will depend on the breadth of its approved labeling and upon the acceptance of the product by the medical community, including physicians, patients and healthcare payors. The degree of market acceptance of any approved product will depend on a number of factors, including, without limitation:
| ● | demonstration of clinical safety and efficacy compared to other products; | |
| ● | ability of physicians to accurately diagnose NASH in its early stages; | |
| ● | the relative convenience and ease of administration; | |
| ● | the prevalence and severity of any adverse side effects; | |
| ● | limitations, warnings or contraindications contained in the product’s approved labeling; | |
| ● | distribution and use restrictions imposed by the FDA, or other regulatory agencies, or agreed to by us as part of a mandatory or voluntary REMS; | |
| ● | availability of alternative treatments, including, any competitive products already approved or expected to be commercially launched in the near future; | |
| ● | pricing and cost effectiveness; | |
| ● | the effectiveness of our, or any future collaborators’, sales and marketing strategies; | |
| ● | our ability to obtain sufficient third-party coverage or reimbursement; and | |
| ● | the willingness of patients to pay for drugs out of pocket in the absence of third-party coverage. |
If a product candidate is approved, but does not achieve an adequate level of acceptance by physicians, healthcare payors and patients, we may not generate sufficient revenue from the product, and we may not become profitable. In addition, our efforts to educate the medical community and third-party payors on the benefits of the product may require significant resources and may never be successful.
| 24 |
The FDA and other regulatory agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses. If we are found to have improperly promoted off-label uses, we may become subject to significant liability.
The FDA and other regulatory agencies strictly regulate the promotional claims that may be made about prescription products. In particular, a product may not be promoted for uses that are inconsistent with the FDA-approved indications and other conditions or restrictions contained in the approved labeling, including the prescribing information, for the product. In particular, any labeling approved by FDA or other foreign regulatory agencies for a product candidate necessarily limits its use for certain conditions in certain patient populations. Also, regulatory agencies may impose further requirements or restrictions on the distribution or use of Aramchol, Amilo-5MER or any other product candidate as part of a mandatory plan, such as limiting prescribing to certain physicians or medical centers that have undergone specialized training, limiting treatment to patients who meet certain safe-use criteria and requiring treated patients to enroll in a registry. If we receive marketing approval for a product candidate, physicians may nevertheless prescribe the product candidate to their patients in a manner that is inconsistent with the approved labeling, which is commonly known as “off label” use. If we are found to have promoted any product candidate for such “off label” uses, we may become subject to significant liability under a variety of statutory theories typically alleged by U.S. regulatory authorities. In particular, the U.S. federal government has levied large civil and criminal fines against companies for alleged improper promotion, has enjoined several companies from engaging in off-label promotion, and has requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed.
Our business and operations may be materially adversely affected in the event of computer system failures or security or breaches due to cyber-attacks or cyber intrusions, including ransomware, phishing attacks and other malicious intrusions.
In recent years, cybersecurity threats have become a greater risk and focus for companies. In particular, ransomware attacks, where a hacker locks and threatens to delete or disclose the victim’s data unless a ransom is paid, has become a major risk. We and those of our CROs and other third parties on which we rely are at risk of cyber-attacks or cyber intrusions via the Internet, computer viruses, break-ins, malware, ransomware, phishing attacks, hacking, denial-of-service attacks or other attacks and similar disruptions from the unauthorized use of, or access to, computer systems (including from internal and external sources). These types of incidents continue to be prevalent and pervasive across industries, including in our industry. In addition, we expect information security risks to continue to increase due to the proliferation of new technologies and the increased sophistication and activities of organized crime, hackers, terrorists and other external parties, including foreign state actors.
Despite the implementation of security measures, our internal computer systems, and those of our CROs and other third parties on which we rely, are vulnerable to damage from computer viruses, unauthorized access, cyber-attacks, cyber intrusions, natural disasters, fire, terrorism, war, and telecommunication and electrical failures. If such an event were to occur and interrupt our operations, it could result in a material disruption of our drug development programs and inflict reputational harm upon us that may result in decreased market value and erode public trust. For example, the loss of clinical trial data from ongoing or planned clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss of or damage to our data or applications, loss of trade secrets or inappropriate disclosure of confidential or proprietary information, including protected health information or personal data of employees or former employees, access to our clinical data, or disruption of the manufacturing process, we could incur liability and the further development of our drug candidates could be delayed.
| 25 |
We may be subject to extensive environmental, health and safety, and other laws and regulations in multiple jurisdictions.
Our business involves the controlled use, through our service providers, of hazardous materials, various biological compounds and chemicals, and as such, we, our agents and our service providers may be subject to various environmental, health and safety laws and regulations, including those governing air emissions, water and wastewater discharges, noise emissions, the use, management and disposal of hazardous, radioactive and biological materials and wastes and the cleanup of contaminated sites. The risk of accidental contamination or injury from these materials cannot be eliminated. If an accident, spill or release of any regulated chemicals or substances occurs, we could be held liable for resulting damages, including for investigation, remediation and monitoring of the contamination, including natural resource damages, the costs of which could be substantial. We may incur substantial capital costs and operating expenses and may be required to obtain consents to comply with any environmental and health laws or regulations and the terms and conditions of any permits required pursuant to such laws and regulations, including costs incurred by us to install new or updated pollution control equipment for our service providers, modify our operations or perform other corrective actions at our facilities or the facilities of our service providers. In addition, fines and penalties may be imposed on us, our agents and/or our service providers for noncompliance with environmental, health and safety and other laws and regulations or for the failure to have, or comply with the terms and conditions of, required environmental or other permits or consents.
We expect the healthcare industry to face increased limitations on reimbursement, rebates and other payments as a result of healthcare reform, which could adversely affect third-party coverage of Aramchol, Amilo-5MER or any other product candidate and how much or under what circumstances healthcare providers will prescribe or administer Aramchol, Amilo-5MER or any other product candidate.
In both the United States and other countries, sales of Aramchol Amilo-5MER or any other product candidate will depend in part upon the availability of reimbursement from third-party payors, which include governmental authorities, managed care organizations and other private health insurers. Third-party payors are increasingly challenging the price and examining the cost effectiveness of medical products and services.
Increasing expenditures for healthcare have been the subject of considerable public attention in the United States. Both private and government entities are seeking ways to reduce or contain healthcare costs. Numerous proposals that would effect changes in the U.S. healthcare system have been introduced or proposed in the U.S. Congress, or Congress, and in some state legislatures, including reducing reimbursement for prescription products and reducing the levels at which consumers and healthcare providers are reimbursed for purchases of pharmaceutical products.
In the United States, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or the Modernization Act, changed the way Medicare covers and pays for most pharmaceutical products in a number of ways. Medicare is the single largest third-party payment program and is administered by the Centers for Medicare & Medicaid Services, or the CMS. Medicare traditionally covered prescription drugs administered by physicians. The Modernization Act introduced a new reimbursement methodology based on average sales prices for many of these drugs. The Modernization Act also established a new competitive acquisition program for the purchase of Part B drugs. This program, when fully implemented, will likely reduce the prices of these drugs. While the Medicare provisions of the Modernization Act apply only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates. Therefore, any reduction in reimbursement that results from federal legislation or regulation may result in a similar reduction in payments from private payors.
Most notably, the Modernization Act also expanded coverage through a new Part D to include ordinary self-administered outpatient drugs. Medicare part D though operates through private insurers, and these insurers negotiate prices with pharmacies and with manufacturers. Intense negotiations can result in reduced revenues to manufacturers.
| 26 |
Increasing expenditures for healthcare have been the subject of considerable public attention in the United States. Both private and government entities are seeking ways to reduce or contain healthcare costs. Numerous proposals that would effect changes in the U.S. healthcare system have been introduced or proposed in U.S. Congress, and in some state legislatures, including reducing reimbursement for prescription products and reducing the levels at which consumers and healthcare providers are reimbursed for purchases of pharmaceutical products.
In March 2010, President Barack Obama signed into law the Patient Protection and Affordable Care Act and the Health Care and Education Affordability Reconciliation Act of 2010, or the Affordable Care Act, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on pharmaceutical and medical device manufacturers and impose additional health policy reforms. The Affordable Care Act expanded manufacturers’ Medicaid rebate liability to include covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations, increased the minimum rebate due for innovator drugs from 15.1% of average manufacturer price, or the AMP, to 23.1% of AMP. The rebate on innovator drugs is the greater of 23.1% of the AMP per unit or the difference between the AMP and the best price per unit and adjusted by the Consumer Price Index-Urban (CPI-U) based on a launch date and current quarter AMP. The total rebate amount for innovator drugs is capped at 100.0% of AMP. The Affordable Care Act and subsequent legislation also narrowed the definition of AMP. Furthermore, the Affordable Care Act imposes a significant annual, nondeductible fee on companies that manufacture or import certain branded prescription drug products. The Affordable Care Act appears likely to continue to put pressure on pharmaceutical pricing, especially under the Medicare and Medicaid programs, and may also increase our regulatory burdens and operating costs.
There have been judicial and congressional challenges to the Affordable Care Act. If a law is enacted, many if not all of the provisions of the PPACA may no longer apply to prescription drugs. While we are unable to predict what changes may ultimately be enacted, to the extent that future changes affect how any future products are paid for and reimbursed by government and private payers our business could be adversely impacted. On December 14, 2018, a federal district court in Texas ruled that the PPACA is unconstitutional as a result of the Tax Cuts and Jobs Act, the federal income tax reform legislation previously passed by Congress and signed by President Trump on December 22, 2017, that eliminated the individual mandate portion of the PPACA. The case, Texas, et al, v. United States of America, et al., (N.D. Texas), is an outlier, and the ruling has been stayed by the ruling judge, but in 2019, the Fifth Circuit Court of Appeals subsequently upheld the lower court decision which was then appealed to the United States Supreme Court. The U.S. Supreme Court declined to hear the appeal on an expedited basis and so no decision is expected until sometime in 2021 before the end of the next Supreme Court’s current term in early 2021. We are not able to state with any certainty what will be the impact of this court decision on our business pending further court action and possible appeals. In November 2020, Joseph Biden was elected President and, in January 2021, the Democratic Party obtained control of the Senate. As a result of these electoral developments, it is unlikely that continued legislative efforts will be pursued to repeal PPACA. Instead, it is possible that executive and regulatory initiatives, as well as legislation will be pursued to enhance or reform PPACA. We are not able to state with certainty what the impact of potential legislation will be on our business.
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. In August 2011, President Obama signed into law the Budget Control Act of 2011, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee did not achieve a targeted deficit reduction of an amount greater than $1.2 trillion for the years 2013 through 2021, triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to healthcare providers of up to 2.0% per fiscal year, starting in 2013. These reductions will stay in effect through 2030 unless additional congressional action is taken. However, COVID-19 relief legislation suspended the 2% Medicare sequester from May 1, 2020 through March 31, 2021. In January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several categories of healthcare providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. If we ever obtain regulatory approval and commercialization of Aramchol or any other product candidate, these laws may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on our customers and accordingly, our financial operations. Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We cannot be sure whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of Aramchol or any other product candidate may be. Further, the Deficit Reduction Act of 2010, directed CMS to contract a vendor to determine “retail survey prices for covered outpatient drugs that represent a nationwide average of consumer purchase prices for such drugs, net of all discounts and rebates (to the extent any information with respect to such discounts and rebates is available).” This survey information can be used to determine the National Average Drug Acquisition Cost, NADAC. Some states have indicated that they will reimburse based on the NADAC and this can result in further reductions in the prices paid for various outpatient drugs.
| 27 |
Various states, such as California, have also taken steps to consider and enact laws or regulations that are intended to increase the visibility of the pricing of pharmaceutical products with the goal of reducing the prices at which pharmaceutical products are sold. Because these various actual and proposed legislative changes are intended to operate on a state-by-state level rather than a national one, we cannot predict what the full effect of these legislative activities may be on our business in the future.
Although we cannot predict the full effect on our business of the implementation of existing legislation or the enactment of additional legislation pursuant to healthcare and other legislative reform, we believe that legislation or regulations that would reduce reimbursement for, or restrict coverage of, Aramchol or any other product candidate, could adversely affect how much or under what circumstances healthcare providers will prescribe or administer our products. This could materially and adversely affect our business by reducing our ability to generate revenue, raise capital, obtain additional collaborators and market Aramchol or any other product candidate. In addition, we believe the increasing emphasis on managed care in the United States has and will continue to put pressure on the price and usage of pharmaceutical products, which may adversely impact any other product sales.
It will be difficult for us to profitably sell any product candidate if reimbursement for the product is limited by government authorities and third-party payor policies.
In addition to any healthcare reform measures that may affect reimbursement, the market acceptance and sales of any product candidate will depend on the reimbursement policies of government authorities and third-party payors. It will be difficult for us to profitably sell a product candidate if reimbursement for the product is limited by government authorities or third-party payors. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will pay for and establish reimbursement levels. A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government authorities and these third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medications. We cannot be sure that coverage or reimbursement will be available for any product candidate and, if coverage and reimbursement are available, of the extent of coverage and the level of reimbursement. Reimbursement may affect the demand for, or the price of, any product for which we obtain marketing approval. In addition, third-party payors are likely to impose strict requirements for reimbursement in order to limit off-label use of a higher priced drug. Reimbursement by a third-party payor may depend upon a number of factors including the third-party payor’s determination that use of a product is:
| ● | a covered benefit under its health plan; | |
| ● | safe, effective and medically necessary; | |
| ● | appropriate for the specific patient; | |
| ● | cost-effective; and | |
| ● | neither experimental nor investigational. |
| 28 |
Obtaining coverage and reimbursement approval for a product from a government or other third-party payor is a time-consuming and costly process that could require us to provide supporting scientific, clinical and cost effectiveness data for the use of a product candidate to the payor. We may not be able to provide data sufficient to gain acceptance with respect to coverage and reimbursement. We cannot be sure that coverage or adequate reimbursement will be available for any product candidates. Also, we cannot be sure that reimbursement amounts will not reduce the demand for, or the price of, any product candidates. If reimbursement is not available, or is available only to limited levels, we may not be able to commercialize any product candidates, profitably, or at all, even if approved. In addition, if physicians, government agencies and other third-party payors do not accept the use or efficacy of any product candidates, we will not be able to generate significant revenue, if any.
Governments outside the United States tend to impose strict price controls, which may adversely affect our revenues, if any.
In some countries, particularly the countries of the EU, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of Aramchol or any other product candidate to other available therapies. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business could be harmed, possibly materially.
If we or any of our independent contractors, consultants, collaborators, manufacturers, or service providers fail to comply with healthcare and data privacy laws and regulations, we or they could be subject to enforcement actions, which could result in penalties and affect our ability to develop, market and sell Aramchol or any other product candidate and may harm our reputation.
We are or may in the future be subject to federal, state, and foreign healthcare and data privacy laws and regulations pertaining to, among other things, fraud and abuse of patients’ rights. These laws and regulations include:
| ● | The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully soliciting, offering, receiving, or paying any remuneration, directly or indirectly, in cash or in kind, to induce or reward purchasing, ordering or arranging for or recommending the purchase or order of any item or service for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid. Liability may be established without a person or entity having actual knowledge of the federal Anti-Kickback Statute or specific intent to violate it. This statute has been interpreted to apply broadly to arrangements between pharmaceutical manufacturers on the one hand and prescribers, patients, purchasers and formulary managers on the other. In addition, the Affordable Care Act amended the Social Security Act to provide that the U.S. government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. A conviction for violation of the Anti-kickback Statute requires mandatory exclusion from participation in federal health care programs. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution, the exemptions and safe harbors are drawn narrowly, and those activities may be subject to scrutiny or penalty if they do not qualify for an exemption or safe harbor. |
| 29 |
| ● | The federal civil False Claims Act, or FCA, prohibits, among other things, knowingly presenting, or causing to be presented claims for payment of government funds that are false or fraudulent, or knowingly making, using or causing to be made or used a false record or statement material to such a false or fraudulent claim, or knowingly concealing or knowingly and improperly avoiding, decreasing, or concealing an obligation to pay money to the federal government. This statute also permits a private individual acting as a “whistleblower” to bring actions on behalf of the federal government alleging violations of the FCA and to share in any monetary recovery. The FCA prohibits anyone from knowingly presenting, conspiring to present, making a false statement in order to present, or causing to be presented, for payment to federal programs (including Medicare and Medicaid) claims for items or services, including drugs, that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services. This law also prohibits anyone from knowingly underpaying an obligation owed to a federal program. Increasingly, U.S. federal agencies are requiring nonmonetary remedial measures, such as corporate integrity agreements in FCA settlements. The U.S. Department of Justice announced in 2016 its intent to follow the “Yates Memo,” taking a far more aggressive approach in pursuing individuals as FCA defendants in addition to the corporations. FCA liability is potentially significant in the healthcare industry because the statute provides for treble damages and mandatory penalties assessed on a per false claim or statement basis. Government enforcement agencies and private whistleblowers have investigated pharmaceutical companies for or asserted liability under the FCA for a variety of alleged promotional and marketing activities, such as providing free product to customers with the expectation that the customers would bill federal programs for the product; providing consulting fees and other benefits to physicians to induce them to prescribe products; engaging in promotion for “off-label” uses; and submitting inflated best price information to the Medicaid Rebate Program. | |
| ● | The federal False Statements Statute prohibits knowingly and willfully falsifying, concealing, or covering up a material fact or making any materially false, fictitious or fraudulent statement or representation, or making or using any false writing or document knowing the same to contain any materially false, fictitious or fraudulent statement or entry, in connection with the delivery of or payment for healthcare benefits, items, or services. | |
| ● | The federal Civil Monetary Penalties Law authorizes the imposition of substantial civil monetary penalties against an entity, such as a pharmaceutical manufacturer, that engages in activities including, among others (1) knowingly presenting, or causing to be presented, a claim for services not provided as claimed or that is otherwise false or fraudulent in any way; (2) arranging for or contracting with an individual or entity that is excluded from participation in federal healthcare programs to provide items or services reimbursable by a federal healthcare program; (3) violations of the federal Anti-Kickback Statute; or (4) failing to report and return a known overpayment. | |
| ● | The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, imposes criminal and civil liability for knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of, or payment for, healthcare benefits, items or services; similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. | |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act (HITECH), which imposes requirements on certain types of people and entities relating to the privacy, security, and transmission of individually identifiable health information, requires notification to affected individuals and regulatory authorities of certain breaches of security of individually identifiable health information. | |
| ● | The federal Physician Payment Sunshine Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program, to report annually to the Centers for Medicare & Medicaid Services (CMS) information related to payments and other transfers of value to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, and ownership and investment interests held by physicians and other healthcare providers and their immediate family members, which is published in a searchable form on an annual basis. Effective January 1, 2022, covered manufacturers will also be required to report on payments and other transfers of value to physician assistants, nurse practitioners or clinical nurse specialists, anesthesiologist assistants, certified registered nurse anesthetists, and certified nurse-midwives during the previous year. |
| 30 |
| ● | State laws comparable to each of the above federal laws, such as, for example, anti-kickback and false claims laws that may be broader in scope and also apply to commercial insurers and other non-federal. | |
| ● | Payors requirements for mandatory corporate regulatory compliance programs, and laws relating to patient data privacy and security. Other state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government; require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state and foreign laws govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. | |
| ● | In the European Union, the General Data Protection Regulation, or GDPR,—Regulation EU 2016/679—was adopted in May 2016 and became applicable on May 25, 2018, or GDPR. The GDPR further harmonizes data protection requirements across the European Union member states by establishing new and expanded operational requirements for entities that collect, process or use personal data generated in the European Union, including consent requirements for disclosing the way personal information will be used, information retention requirements, and notification requirements in the event of a data breach. | |
| ● | In the United Kingdom, following the UK’s exit from the European Union on 31 December 2020, the GDPR continues to form part of the law in the UK with some amendments (UK GDPR). There is a risk of divergence in the future, which may increase our overall data protection compliance costs. | |
| ● | The California Consumer Privacy Act of 2018, or CCPA, effective as of January 1, 2020, gives California residents expanded rights to access and require deletion of their personal information, opt out of certain personal information sharing, and receive detailed information about how their personal information is used. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches, that is expected to increase data breach litigation. | |
| ● | In addition, failure to comply with the Israeli Privacy Protection Law of 1981, and its regulations, as well as the guidelines of the Israeli Privacy Protection Authority, may expose us to administrative fines, civil claims (including class actions) and in certain cases criminal liability. Current pending legislation may result in a change of the current enforcement measures and sanctions. |
If our operations are found to be in violation of any such health care laws and regulations, we may be subject to penalties, including significant administrative, civil and criminal penalties, monetary damages, disgorgement, imprisonment, the curtailment or restructuring of our operations, loss of eligibility to obtain approvals from the FDA or foreign regulatory authorities, or exclusion from participation in government contracting, healthcare reimbursement or other government programs, including Medicare and Medicaid, any of which could adversely our financial results. Any action against us for an alleged or suspected violation could cause us to incur significant legal expenses and could divert our management’s attention from the operation of our business, even if our defense is successful. In addition, achieving and sustaining compliance with applicable laws and regulations may be costly to us in terms of money, time and resources.
| 31 |
Our employees, principal investigators, consultants, commercial partners or vendors may engage in misconduct or other improper activities, including non-compliance with regulatory standards.
We are also exposed to the risk of employees, independent contractors, principal investigators, consultants, commercial partners or vendors engaging in fraud or other misconduct. Misconduct by employees, independent contractors, principal investigators, consultants, commercial partners and vendors could include intentional failures to comply with EU or UK regulations, to provide accurate information to the EMA, MHRA or EU Member States authorities or to comply with manufacturing or quality standards we have or will have established. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices such as promotion of products by medical practitioners. The EU Member States in which we operate have different statutory provisions regulating the cooperation of pharmaceutical companies with healthcare professionals. In addition to these statutory provisions, codes of conduct issued by business associations or other non-statutory standards may be applicable to our activities. Both statutory provisions and non-statutory codes or standards restrict payments or other benefits provided to healthcare professionals, and in case of non-compliance, may result in severe sanctions such as bans, administrative fines, criminal fines or even imprisonment. The advertising of medicinal products for human use in the EU is regulated by Title VIII of European Directive 2001/83/EC. These provisions have been implemented into the law of the EU member States. Such laws inter alia restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Misconduct could also involve the improper use of information obtained in the course of clinical studies, which could result in regulatory sanctions and serious and irreparable harm to our reputation.
This could also apply with respect to data privacy. In the EU and United Kingdom, the EU Directive 95/46/EEC was replaced by the GDPR on May 25, 2018. The GDPR as an EU regulation does not have to be implemented into Member States’ national law, but applies directly in all Member States since May 25, 2018. It applies to companies with an establishment in the European Economic Area (EEA) and to certain other companies not in the EEA that offer or provide goods or services to individuals located in the EEA or monitor individuals located in the EEA. The GDPR implements more stringent operational requirements for controllers of personal data, including, for example, expanded disclosures about how personal information is to be used, limitations on retention of information, increased requirements pertaining to health data and pseudonymized (i.e., key-coded) data, increased cyber security requirements, mandatory data breach notification requirements and higher standards for controllers to demonstrate that they have obtained a valid legal basis for certain data processing activities. The GDPR provides that EU Member States may continue to make their own further laws and regulations in relation to the processing of genetic, biometric or health data, which could result in continued or new differences between Member States, limit our ability to use and share personal data or could cause our costs to increase, and harm our business and financial condition. The GDPR continues to form part of law in the United Kingdom with some amendments following the United Kingdom’s exit from the European Union on 31 December 2020 (UK GDPR) although there is a risk of divergence in the future which may increase our overall data protection compliance cost. We are also subject to evolving and strict rules on the transfer of personal data out of the European Union and United Kingdom to the United States. Further prospective revision of the Directive on privacy and electronic communications (Directive 2002/58/EC), or ePrivacy Directive, and equivalent United Kingdom Legislation may affect our marketing communications.
We have implemented procedures to ensure compliance with the GDPR and UK GDPR and its requirements. Our actual or alleged failure to comply with this regulation, or to protect personal data, could result in enforcement actions and significant penalties against us, which could result in negative publicity, increase our operating costs, subject us to claims or other remedies and have a material adverse effect on our business, financial condition, and results of operations. It is not always possible to identify and deter misconduct by employees or other parties. The precautions we take to detect and prevent such activity may not protect us from legal or regulatory action resulting from a failure to comply with applicable laws or regulations. Misconduct by our employees, principal investigators, consultants, commercial partners or vendors could result in significant financial penalties, criminal sanctions, civil law claims and/or negative media coverage, and thus have a material adverse effect on our business, including through the imposition of significant fines or other sanctions, and our reputation. In particular, failure to comply with EU laws, including failure under the GDPR, UK GDPR, ePrivacy Directive and other laws relating to the security of personal data may result in fines up to €20,000,000 or up to 4% of the total worldwide annual turnover of the preceding financial year, if greater, and other administrative penalties including criminal liability, which may be onerous and adversely affect our business, financial condition, results of operations and prospects. Failure to comply with the GDPR, UK GDPR and related laws may also give risk to increase risk of private actions, including a new form of class action that is available under the GDPR and UK GDPR.
| 32 |
If we or our manufacturers fail to comply with manufacturing regulations, our financial results and financial condition could be adversely affected.
Before an NDA is approved, and before we begin the commercial manufacture of any product candidate, contract manufacturers must register with FDA or foreign regulators undergo regulatory inspection of their manufacturing facilities, processes and quality systems. In addition, pharmaceutical manufacturing facilities are subject to periodic inspection by the FDA and foreign regulatory authorities after product approval. Due to the complexity of the processes used to manufacture pharmaceutical products and product candidates, any potential third-party manufacturer may be unable to meet local, federal, or international regulatory requirements either at the outset or on an ongoing basis, in a cost-effective manner, if at all.
We do not intend to engage in the manufacture of Aramchol or any other product candidate other than for pre-clinical and clinical studies, but we or our materials suppliers may face manufacturing or quality control problems causing product production and shipment delays or a situation where we or the supplier may not be able to maintain compliance with the FDA’s or foreign regulators’ requirements necessary to continue manufacturing Aramchol or any other product candidate. Drug manufacturers are subject to ongoing periodic unannounced inspections by the FDA and corresponding foreign regulators to ensure continuing compliance with applicable requirements. Any failure to comply with FDA or foreign regulatory requirements could adversely affect our clinical research activities and our ability to develop and market Aramchol or any other product candidate.
If a third-party manufacturer with whom we contract is unable to comply with manufacturing requirements, we may be subject to fines, unanticipated compliance expenses, recall or seizure of Aramchol or any other product candidate, total or partial suspension of production and/or enforcement actions, including injunctions, and criminal or civil prosecution. These possible sanctions could adversely affect our financial results and financial condition.
Our market is subject to intense competition. If we are unable to compete effectively, Aramchol, Amilo-5MER or any other product candidate that we develop may be rendered suboptimal, noncompetitive or obsolete.
There are a number of products in development for our target indications, many of which are being developed by pharmaceutical companies that are far larger than us, with significantly greater resources and more experience than us in all aspects of drug development and commercialization. Further, our industry is highly competitive and subject to rapid and significant technological change. Our potential competitors include large, fully-integrated pharmaceutical and biotechnology companies, specialty pharmaceutical and generic drug companies, academic institutions, government agencies and research institutions. All of these competitors currently engage in, have engaged in or may engage in the future in the development, manufacturing, marketing and commercialization of new pharmaceuticals, some of which may compete with Aramchol, Amilo-5MER or other product candidates. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. These companies may have products in development that are superior to any Aramchol or any other product candidate. Key competitive factors affecting the commercial success of Aramchol or any other product candidate that we develop are likely to be efficacy, time of onset, safety and tolerability profile, reliability, convenience of dosing, price and reimbursement.
| 33 |
Many of our potential competitors have substantially greater financial, technical and human resources than we do and significantly greater experience in the discovery and development of drug candidates, obtaining FDA and other regulatory approvals of products and the commercialization of those products. Accordingly, our competitors may be more successful than us in obtaining FDA and other marketing approvals for drugs and achieving widespread market acceptance. Our competitors’ drugs may be more effective, or more effectively marketed and sold, than any drug we may commercialize and may render the product candidates that we develop suboptimal, obsolete or non-competitive before we can recover the expenses of developing and commercializing the product. We anticipate that we will face intense and increasing competition as new drugs enter the market and advanced technologies become available. Finally, the development of new treatment methods for the diseases we are targeting could render Aramchol or any other product candidate that we develop, non-competitive or obsolete. If we cannot successfully compete with new or existing products, our marketing and sales will suffer and we may never be profitable.
Our competitors currently include companies with marketed products and/or advanced clinical programs. Our main competitors of Aramchol include, but are not limited to Novo Nordisk, Intercept Pharmaceuticals, Inc., Inc., Madrigal Pharmaceuticals Inc., and Viking Therapeutics among others. See also “Item 4. Information on the Company—Competition.” Moreover, several additional companies have reported the commencement of research projects and proof-of-concept trials related to our target indications, including those mentioned in the preceding sentence
We face potential product and other liability exposure, and, if claims are brought against us, we may incur substantial liability.
Our product candidates could cause adverse events. These adverse events may not be observed in clinical trials, but may nonetheless occur in the future. If any of these adverse events occur, they may render Aramchol or any other product candidate ineffective or harmful in some patients, and our sales would suffer, materially adversely affecting our business, financial conditions and results of operations.
In addition, potential adverse events caused by Aramchol or any other product candidate, could lead to product liability claims. Product liability claims might be brought against us by consumers, healthcare providers or others coming into contact with Aramchol or any other product candidate. If we cannot successfully defend ourselves against product liability claims, we could incur substantial liabilities. In addition, regardless of merit or eventual outcome, product liability claims may result in, among other things:
| ● | decreased demand for our product candidate for which we obtain marketing approval; | |
| ● | impairment of our business reputation and exposure to adverse publicity; | |
| ● | increased warnings on product labels or other regulatory actions; | |
| ● | withdrawal of clinical trial participants; | |
| ● | costs of related litigation; | |
| ● | distraction of management’s attention from our primary business; | |
| ● | substantial monetary awards to patients or other claimants; | |
| ● | loss of revenue; and | |
| ● | the inability to successfully commercialize any product candidates, for which we obtain marketing approval. |
| 34 |
If we are unable to obtain adequate insurance with respect to our clinical trials against and from any losses or claims from third parties, our financial condition could be adversely affected in the event of uninsured or inadequately insured loss or damage. We may not be able to obtain insurance policies on terms affordable to us that would adequately cover loss or claims by third parties. To the extent our business suffers any losses or claims by third parties, which are not covered, or adequately covered, by insurance, our financial condition may be materially adversely affected.
If product liability lawsuits are successfully brought against us, our insurance may be inadequate.
We have obtained insurance coverage for our clinical trials in accordance with market standards and in compliance with applicable Israeli law. However, our insurance coverage may not be sufficient to reimburse us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive, and, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. If and when we obtain marketing approval for any product candidate, we intend to expand our insurance coverage to include the sale of commercial products; however, we may be unable to obtain this product liability insurance on commercially reasonable terms. On occasion, large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. The cost of any product liability litigation or other proceedings, even if resolved in our favor, could be substantial. A successful product liability claim, or series of claims, brought against us could cause our share price to decline and, if judgments exceed our insurance coverage, could decrease our cash and adversely affect our business.
The product liability insurance we will need to obtain in connection with the commercial sales of any product candidate, if and when they receive regulatory approval, may be unavailable in meaningful amounts or at a reasonable cost. If we are the subject of a successful product liability claim that exceeds the limits of any insurance coverage we obtain, we would incur substantial charges that would adversely affect our earnings and require the commitment of capital resources that might otherwise be available for the development and commercial launch of any product candidate’s programs.
We manage our business through a small number of senior executive officers. We depend on them even more than similarly- situated companies.
Because of the specialized scientific and managerial nature of our business, we rely heavily on our ability to recruit, attract, retain, manage and motivate qualified senior executive officers with adequate operational, scientific and technical experience. The loss of the services of our senior executive officers, including our President, Chief Executive Officer, and our Chief Scientific Officer, or the inability to hire or retain experienced management personnel, could adversely affect our ability to execute our business plan and harm our operating results. In particular, the loss of one or more of our senior executive officers could be detrimental to us if we cannot recruit suitable replacements in a timely manner.
We do not currently carry “key person” insurance on the lives of members of senior management. The competition for qualified personnel in the pharmaceutical field is intense. Due to this intense competition, we may be unable to attract and retain qualified personnel necessary for the development of our business or to recruit suitable replacement personnel. Additionally, our ability to effectively recruit and retain qualified officers and directors could also be adversely affected if we experience difficulty in obtaining adequate directors’ and officers’ liability insurance. We may be unable to maintain sufficient insurance as a public company to cover liability claims made against our officers and directors. If we are unable to adequately insure our officers and directors, we may not be able to retain or recruit qualified officers and directors to manage the Company.
| 35 |
Failure to build our finance infrastructure and improve our accounting systems and controls could impair our ability to comply with the financial reporting and internal control requirements for publicly traded companies.
As a public company, we operate in an increasingly challenging regulatory environment which requires us to comply with the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, and the related rules and regulations of the SEC and securities exchanges, expanded disclosures, accelerated reporting requirements and more complex accounting rules. Company responsibilities required by the Sarbanes-Oxley Act include establishing corporate oversight and adequate internal control over financial reporting and disclosure controls and procedures. Effective internal controls are necessary for us to produce reliable financial reports and are important to help prevent financial fraud.
Section 404 of the Sarbanes-Oxley Act requires our management to report on, and our independent registered public accounting firm to attest to, the effectiveness of our internal control structure and procedures for financial reporting. We have an ongoing program to perform the system and process evaluation and testing necessary to continue to comply with these requirements. During the course of our review and testing, we may identify deficiencies and be unable to remediate them before we must provide the required reports. Furthermore, if we have a material weakness in our internal controls over financial reporting, we may not detect errors on a timely basis and our financial statements may be materially misstated. We or our independent registered public accounting firm may not be able to conclude on an ongoing basis that we have effective internal control over financial reporting, which could harm our operating results, cause investors to lose confidence in our reported financial information and cause the trading price of our stock to fall.
To build our finance infrastructure, we may need to improve our accounting systems, disclosure policies, procedures and controls. If we are unsuccessful in building an appropriate accounting infrastructure, we may not be able to prepare and disclose, in a timely manner, our financial statements and other required disclosures, or comply with existing or new reporting requirements. Any failure to report our financial results on an accurate and timely basis could result in sanctions, lawsuits, delisting of our shares from the Nasdaq Capital Market or other adverse consequences that would materially harm our business. If we cannot provide reliable financial reports or prevent fraud, our business and results of operations could be harmed and investors could lose confidence in our reported financial information.
We may need to significantly increase the size of our organization, and we may experience difficulties in managing growth.
We may experience rapid and substantial growth in order to achieve our operating plans, which will place a strain on our human and capital resources. Successful implementation of our business plan will require management of growth, which will result in an increase in the level of responsibility for management personnel. Any future growth will impose significant added responsibilities on members of management, including the need to identify, recruit, maintain and integrate additional employees. To that end, we must be able to, among other things:
| ● | manage our clinical trials and the regulatory process effectively; | |
| ● | develop our administrative, accounting and management information systems and controls; | |
| ● | hire and train additional qualified personnel; and | |
| ● | integrate current and additional management, administrative, financial and sales and marketing personnel. |
If we are unable to establish, scale-up and implement improvements to our control systems in an efficient or timely manner, or if we encounter deficiencies in existing systems and controls, investors may choose not to invest in us, which could cause our share price to decline and negatively impact our ability to successfully commercialize Aramchol or any other product candidate.
| 36 |
Failure to attract and retain sufficient numbers of talented employees will further strain our human resources and could impede our growth or result in ineffective growth. If we are unable to manage our growth effectively, our losses could materially increase and it will have a material adverse effect on our business, results of operations and financial condition.
Our business, including our ability to raise capital, may be affected by macroeconomic conditions.
A deterioration in global economic conditions and uncertainties may have an adverse effect on our business. For instance, interest rates, the liquidity of the credit markets and the volatility of the capital markets could also affect the value of our investments, if any, and our ability to liquidate such investments in order to fund our operations. Interest rates and the ability to access credit markets could also adversely affect the ability of patients and distributors to purchase, pay for and effectively distribute Aramchol or any other product candidate.
In addition, we rely and intend to rely on third-parties, including our clinical research organizations, third-party manufacturers and second source suppliers, and certain other important vendors and consultants. As a result of volatile and unpredictable global economic situations, there may be a disruption or delay in the performance of our third-party contractors and suppliers. If such third-parties are unable to satisfy their contractual commitments to us, our business could be severely adversely affected.
Additional clinical trials may divert a significant amount of our resources and may ultimately be unsuccessful.
We are seeking to expand our clinical operations for Aramchol to multiple other indications in order to expand our pipeline, commercial potential and ultimately de-risk the Company for the success of any one given trial. If we initiate additional clinical trials, this may divert a significant amount of Company resources and may be unsuccessful. The regulatory pathway may prove simpler than for Nash and as a result this may shift the priorities for Aramchol in the future.
Our business is subject to risks arising from epidemic diseases, such as the recent COVID-19 pandemic, which has impacted and could continue to impact our business.
In late 2019, a novel strain of COVID-19, also known as coronavirus, was reported in Wuhan, China. Initially the outbreak was largely concentrated in China, but it rapidly spread to countries across the globe, including in Israel and the United States. Many countries around the world, including Israel and the United States, implemented significant governmental measures to control the spread of the virus, including temporary closure of businesses, severe restrictions on travel and the movement of people, and other material limitations on the conduct of business. In response, we implemented remote working and workplace protocols for our employees in accordance with Israeli Ministry of Health requirements to ensure employees safety. Many of our trial sites in our ARMOR Study are based in areas currently affected by COVID-19 and there is a general unease of conducting scheduled or elective procedures in medical centers. Given the significant strains on the healthcare system across the globe, during 2020 we temporarily halted the screening of new patients for the ARMOR Study and temporarily suspended the opening of new trial sites. Although we subsequently resumed screening activities and recruitment, in December 2020 we announced the addition of an open label part to the ARMOR Study and suspended randomization of new patients into the double-blind, placebo-controlled histology-based registrational phase of the ARMOR Study as currently enrolled patients are transitioned to the open label part. The open label part of the ARMOR Study is being conducted in a smaller subset of the ARMOR study sites which have been less affected by the COVID-19 pandemic. We continue to closely monitor the local situation in the U.S. and other countries around the world. To help mitigate cost overrun, during 2020 and 2021 we took several cost reduction measures including minimizing clinical related expenses, making certain adjustments to clinical staff and pay according to the current and predicted level of activity, and we downsized our in-house clinical force.
| 37 |
In addition, the rapid development and fluidity of the COVID-19 pandemic precludes any firm estimates as to the ultimate effect this disease will have on our clinical trials, our operations and our business and it is not possible to predict the impact of the second and any further wave of COVID-19. As a result, any current assessment of the effects of the COVID-19 pandemic, including the impact of this disease on the ARMOR Study and any other pre-clinical or clinical studies, is difficult to predict and subject to change and we may experience further disruptions that could severely impact our business, clinical trials, and supply chains, including:
| ● | interruption of key clinical trial activities, such as clinical trial site monitoring, due to limitations on travel imposed or recommended by federal or state governments, employers and others or interruption of clinical trial subject visits and study procedures, which may impact the integrity of subject data and clinical study endpoints; | |
| ● | interruption of, or delays in receiving, supplies of Aramchol or any other product candidate from our contract manufacturing organizations due to staffing shortages, production slowdowns or stoppages and disruptions in delivery systems; | |
| ● | diversion of healthcare resources away from the conduct of clinical trials, including the diversion of hospitals and other medical centers serving as our clinical trial sites and hospital and other staff supporting the conduct of our clinical trials; | |
| ● | delays or difficulties in clinical site initiation, including difficulties in recruiting clinical site investigators and clinical site staff for the ARMOR Study or any other clinical trial; | |
| ● | delays or difficulties in enrolling patients for the ARMOR Study or any other clinical trial especially if sites do not reopen to screen and enroll patients; | |
| ● | delays in clinical sites receiving the supplies and materials needed to conduct the ARMOR Study or any other clinical trial and interruption in global shipping that may affect the transport of clinical trial materials; | |
| ● | limitations on employee resources that would otherwise be focused on the conduct of the ARMOR Study or any other clinical trial, including because of sickness of employees or their families or the desire of employees to avoid contact with large groups of people; | |
| ● | interruptions or delays in the operations of the FDA, EMA, MHRA or other regulatory authorities, including in receiving feedback or approvals from the FDA, EMA, MHRA or other regulatory authorities with respect to regulatory submissions; | |
| ● | changes in local regulations as part of a response to COVID-19 which may require us to change the ways in which the ARMOR Study or any other clinical trial is being conducted, which may result in unexpected costs, or to discontinue the clinical trials altogether; | |
| ● | delays in necessary interactions with local regulators, ethics committees and other important agencies and contractors due to limitations in employee resources or forced furlough of government employees; | |
| ● | refusal of the FDA, EMA, MHRA or other regulatory authorities to accept data from clinical trials in affected geographies; and | |
| ● | impacts from prolonged remote work arrangements, such as increased cybersecurity risks and strains on our business continuity plans. |
| 38 |
In addition, the spread of COVID-19 has had and may in the future impact the trading price of shares of our ordinary shares and could impact our ability to raise additional capital on a timely basis or at all. The COVID-19 pandemic continues to evolve. The extent to which the COVID-19 pandemic may impact our operations will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the geographic spread of the disease, the duration of the pandemic, travel restrictions, quarantines, shelter-in-place orders and social distancing, business closures or business disruptions and the effectiveness of actions taken to contain and treat the disease. The impact of the COVID-19 pandemic may also have the effect of heightening many of the other risks described in the “Risk Factors” section of this Annual Report on Form 20-F.
Risks Related to Our Reliance on Third Parties
We have no manufacturing capacity and anticipate reliance on third-party manufacturers for Aramchol, Amilo-5MER or any other product candidates.
We do not currently operate manufacturing facilities for the production of Aramchol, Amilo-5MER or their API. We still have not, and may never, develop facilities for the manufacture of product candidates or products for clinical trials or commercial purposes. We rely, and for the foreseeable future, will continue to rely, on third-party manufacturers to produce bulk drug products required for our clinical trials. We plan to initially rely upon contract manufacturers and, potentially, collaboration partners, to manufacture commercial quantities of Aramchol or any other product candidate, if and when approved for marketing by the applicable regulatory authorities. Our contract manufacturers have not completed commercial development and validation processes of the Aramchol and Amilo-5Mer API and we have experienced delays in the development of the Aramchol meglumine formulation. Drug product manufacturing processes for clinical batches is still under development and may experience further difficulties and delays in production.
Additionally, if our contract manufacturers and their facilities, as applicable, are not approved by the FDA, or other applicable regulatory authorities, our commercial supply of the drug substance will be significantly delayed and may result in significant additional costs. We purchase finished Aramchol from a third-party under a clinical supply agreement. If we will be required to change the finished product manufacturer, we may encounter significant delay and likely significant additional cost.
A failure by our contract manufacturer to achieve and maintain high manufacturing standards, in accordance with applicable good manufacturing practices and other applicable regulatory requirements could result in patient injury or death, product shortages, product recalls or withdrawals, delays or failures in product testing or delivery, cost overruns or other problems that could seriously harm our business. Contract manufacturers often encounter difficulties involving production yields, quality control and quality assurance, as well as shortages of qualified personnel.
Our existing manufacturers and any future contract manufacturers may not perform as agreed or may not remain in the contract manufacturing business. In the event of a natural disaster, business failure, strike or other difficulty, we may be unable to replace a third-party manufacturer in a timely manner and the production of our product candidates would be interrupted, resulting in delays and additional costs.
We intend to rely primarily on third parties to market and sell Aramchol, Amilo-5MER or any other product candidate.
We have no sales or distribution capabilities. To the extent we rely on third parties to commercialize Aramchol, Amilo-5MER or any other product candidate, if marketing approval is obtained, we may receive less revenue than if we commercialize them ourselves. In addition, we would have less control over the sales efforts of any third parties involved in our commercialization efforts. In the event we are unable to collaborate with a third-party marketing and sales organization to commercialize Aramchol, Amilo-5MER or any other product candidate, particularly for broader patient populations, our ability to generate revenue will be limited.
| 39 |
Although we may ultimately develop a marketing and sales force with technical expertise and supporting distribution capabilities in the longer term, we do not currently intend to do so and, as such, we will be unable to market our product candidates directly in the near future. To promote any of our potential products through third parties, we will have to locate acceptable third parties for these functions and enter into agreements with them on acceptable terms, and we may not be able to do so. Any third-party arrangements we are able to enter into may result in lower revenues than we could achieve by directly marketing and selling our potential products. In addition, to the extent that we depend on third parties for marketing and distribution, any revenues we receive will depend upon the efforts of such third parties, as well as the terms of our agreements with such third parties, which cannot be predicted in most cases at this time. As a result, we might not be able to market and sell our product candidates in the United States or overseas, which would have a material adverse effect on us.
Any collaboration arrangements that we may enter into in the future may not be successful, which could adversely affect our ability to develop and commercialize our current and potential other product candidates.
We intend to seek collaboration arrangements with pharmaceutical or biotechnology companies for the continued development and commercialization of our current and potential other product candidates. We will face, to the extent that we decide to enter into collaboration agreements, significant competition in seeking appropriate collaborators. Moreover, collaboration arrangements are complex and time consuming to negotiate, document and implement. We may not be successful in our efforts to establish and implement collaborations or other alternative arrangements. The terms of any collaborations or other arrangements that we may establish may not be favorable to us.
Any future collaborations that we enter into may not be successful. The success of our collaboration arrangements will depend heavily on the efforts and activities of our collaborators. Collaborators generally have significant discretion in determining the efforts and resources that they will apply to these collaborations. Disagreements between parties to a collaboration arrangement regarding clinical development and commercialization matters can lead to delays in the development process or commercializing the applicable product candidate and, in some cases, termination of the collaboration arrangement. These disagreements can be difficult to resolve if neither of the parties has final decision making authority. Moreover, collaborations with pharmaceutical or biotechnology companies and other third parties are often terminated or allowed to expire by the other party. Any lack of effort or ability by our collaborators or any such disagreement, termination or expiration could adversely affect us financially and could harm our business reputation.
We depend on third parties to conduct our clinical trials.
We rely on third parties, such as contract research organizations, medical institutions, clinical investigators and contract laboratories to oversee most of the operations of our clinical trials and to perform data collection and analysis. As a result, we may face additional delays outside of our control if these parties do not perform their obligations in a timely fashion or in accordance with regulatory requirements. If these third parties do not successfully carry out their contractual duties or obligations and meet expected deadlines, if they need to be replaced, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or for other reasons, our financial results and the commercial prospects for Aramchol, Amilo-5MER or any other product candidate could be harmed, our costs could increase and our ability to obtain regulatory approval and commence product sales could be delayed.
| 40 |
Risks Related to Our Intellectual Property
The failure to obtain or maintain patents, licensing agreements and other intellectual property rights that are sufficiently broad and protective could impact our ability to compete effectively.
To compete effectively, we must develop and maintain a proprietary position with regard to our own technologies, intellectual property, licensing agreements, product candidates and business. Legal standards relating to the validity and scope of claims in the biotechnology and biopharmaceutical fields are still evolving. We cannot predict the scope and extent of patent protection for Aramchol, Amilo-5MER or any other product candidate because the patent positions of pharmaceutical products are complex and uncertain. Therefore, the degree of future protection for our proprietary rights in our core technologies and any product candidates or products that might be developed using these technologies is also uncertain. The risks and uncertainties that we face with respect to our patents and other proprietary rights include, but are not limited to, the following:
| ● | while the patents we own have been issued, pending patent applications we have filed may not result in issued patents or may take longer than we expect to result in issued patents; | |
| ● | we may be subject to interference, reexamination, inter pares review, or post-grant review proceedings in the U.S.; | |
| ● | we may be subject to opposition proceedings in certain foreign countries; | |
| ● | any patents that are issued may not provide meaningful protection for any significant period of time, if at all; | |
| ● | any issued patents may not be broad or strong enough to prevent competition from other products including identical or similar products; | |
| ● | we may not be able to develop additional proprietary technologies that are patentable; | |
| ● | there may be prior art of which we are not aware that may affect the validity or enforceability of a patent claim; | |
| ● | there may be other patents or pending patent applications existing in the patent landscape that will affect our freedom to operate for our product candidates; | |
| ● | other companies may challenge and invalidate patents licensed or issued to us or our customers; | |
| ● | a court could determine that a competitor’s technology or product does not infringe our patents; | |
| ● | other companies may independently develop similar or alternative technologies, or duplicate our technologies; | |
| ● | other companies may design around technologies we have licensed or developed; | |
| ● | if we are not awarded patents or if issued patents expire or are declared invalid or not infringed, there may be no protections against competitors making generic equivalents; | |
| ● | enforcement of patents is complex, uncertain and expensive, and our patents may be found invalid or enforceable; | |
| ● | our patents could irretrievably lapse due to failure to pay fees or otherwise comply with regulations, or could be subject to compulsory licensing; and | |
| ● | if we encounter delays in our development or clinical trials, the period of time during which we could market our product candidates under patent protection would be reduced. |
| 41 |
We cannot be certain that patents will be issued as a result of any of our pending applications, and we cannot be certain that any of our issued patents, whether issued pursuant to our pending applications or licensed from third parties, will give us adequate protection from competing products. For example, issued patents may be circumvented or challenged, declared invalid or unenforceable, or narrowed in scope. In addition, because publication of discoveries in the scientific or patent literature often lags behind actual discoveries, we cannot be certain that we were the first to make our inventions or to file patent applications covering those inventions. If any of our composition of matter patents, or pending applications, was subject to a successful challenge or failed to issue, our business and competitive advantage could be significantly affected. Our current patents will expire or they may otherwise cease to provide meaningful competitive advantage, and we may be unable to adequately develop new technologies and obtain future patent protection to preserve our competitive advantage or avoid adverse effects on our business.
We are currently planning to transition from Aramchol free acid to Aramchol meglumine (salt) in our double-blind, placebo-controlled histology-based registrational part of the ARMOR Study. We had originally been working towards submission of a new drug application, or NDA, in the first half of 2023, assuming positive top-line results, however due to delays in enrollment of the ARMOR Study, we will not be able to submit an NDA to the FDA with respect to Aramchol free acid, a new chemical entity, in time to benefit from any potential Hatch-Waxman patent restoration term. As part of our research and development studies, we have confirmed that several Aramchol salts have improved solubility as compared to the existing form of Aramchol free acid. We have pending patent applications and have been granted patents directed to composition of matter of Aramchol meglumine as well as a wide range of other salts; a method for treating/ inhibiting hepatic fibrosis and non-hepatic fibrosis associated or non-associated with non-alcoholic fatty acid liver disease; and a method of treating dysbiosis. In addition, we have since submitted additional patent applications for Aramchol meglumine and other salts, including a low dose composition for Aramchol meglumine and other salts. We have since been granted a composition of matter patents for Aramchol meglumine and other salts which includes claims for the treatment of fatty liver in Europe and certain other countries while the composition of matter patent application is still pending in the U.S. and certain other countries (India and Brazil) and we have been granted a low dose composition of matter patent for Aramchol meglumine in the U.S.
There can be no assurance that the U.S. Patent and Trademark Office, or the USPTO, or any other foreign equivalent will issue any additional patents based on the patent applications that we submitted to protect our Aramchol salts or Amilo-5MER, nor, should the USPTO or foreign equivalent issue any patents to us with respect to the Aramchol salts or Amilo-5MER, that we will be provided with adequate protection against potentially competitive products. Furthermore, if the USPTO or foreign equivalent issues us one or more patents for the Aramchol salts or Amilo-5MER or with respect to already issued patents for the Aramchol salts or Amilo-5MER, there can be no assurance that the issued patents will be of any commercial value, or that private parties or competitors will not successfully challenge these patents or circumvent these patents in the United States or their counterparts abroad. In the absence of adequate patent protection, our business may be adversely affected by competitors who develop comparable technology or products and our commercial prospects may be materially adversely affected.
Others may obtain issued patents that could prevent us from commercializing our product candidates or require us to obtain licenses requiring the payment of significant fees or royalties in order to enable us to conduct our business. As to those patents that we have licensed, our rights depend on maintaining our obligations to the licensor under the applicable license agreement, and we may be unable to do so.
In addition to patents and patent applications, we depend upon trade secrets and proprietary know-how to protect our proprietary technology. We require our employees, consultants, advisors and collaborators to enter into confidentiality agreements that prohibit the disclosure of confidential information to any other parties. We also require our employees and consultants to disclose and assign to us their ideas, developments, discoveries and inventions. These agreements may not, however, provide adequate protection for our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure.
| 42 |
Patent terms may be inadequate to protect our competitive position on our product candidates for an adequate amount of time.
Patents have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional filing date. Various extensions may be available, but the life of a patent, and the protection it affords, is limited. Even if patents covering our product candidates are obtained, once the patent life has expired, we may be open to competition from competitive products, including generics or biosimilars. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
We may not be able to enforce our intellectual property rights throughout the world. This risk is exacerbated for us because we expect Aramchol or Aramchol meglumine and Amilo-5MER will be manufactured and used in a number of foreign countries.
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. This risk is exacerbated for us because we expect Aramchol or Amilo-5MER will be manufactured and used in a number of foreign countries.
The legal systems of some countries, particularly developing countries, do not favor the enforcement of patents and other intellectual property protection, especially those relating to life sciences. This could make it difficult for us to stop the infringement of our other intellectual property rights. For example, several foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, some countries limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, patents may provide limited or no benefit.
Although most jurisdictions in which the Company has applied for, intends to apply for, or has been issued patents have patent protection laws similar to those of the United States, some of them do not. For example, the Company expects to do business in South America, Eurasia, China and Indochina in the future and the countries in these regions may not provide the same or similar protection as that provided in the United States.
Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate. In addition, changes in the law and legal decisions by courts in the United States and foreign countries may affect our ability to obtain adequate protection for our technology and the enforcement of intellectual property.
We may rely on third party patents.
We may not have rights under some patents or patent applications related to products we may commercialize in the future. Third parties may own or control these patents and patent applications in the United States and abroad. Therefore, in some cases, to manufacture, sell or import some of our future products, we or our collaborators may choose to seek, or be required to seek, licenses under third party patents issued in the United States and abroad or under patents that might be issued from United States and foreign patent applications. In instances in which we must obtain a license for third party patents, we may be required to pay license fees or royalties or both to the licensor. If licenses are not available to us on acceptable terms, we or our collaborators may not be able to develop, manufacture, sell or import these products.
| 43 |
We may be unable to protect the intellectual property rights of third parties from whom we may license certain of our intellectual property or with whom we have entered into other strategic relationships, which could have a material adverse effect on our business, results of operations and financial condition.
Certain of our intellectual property rights may be licensed from third parties, including universities and strategic partners. Such third parties may determine not to or fail to protect the intellectual property rights that we license from them and we may be unable to defend such intellectual property rights on our own or we may have to undertake costly litigation to defend the intellectual property rights of such third parties. There can be no assurances that we will continue to have proprietary rights to any of the intellectual property that we license from such third parties or otherwise have the right to use through similar strategic relationships. Any loss or limitations on use with respect to such intellectual property licensed from third parties or otherwise obtained from third parties with whom we have entered into strategic relationships could have a material adverse effect on our business, results of operations and financial condition.
If we fail to comply with our obligations in the agreements under which we license intellectual property rights from third parties or otherwise experience disruptions to our business relationships with our licensors, we could lose intellectual property rights that are important to our business.
We may be party to license agreements with third parties and may need to obtain additional licenses from others to advance our research and development activities or allow the commercialization product candidates we may identify and pursue. License agreements may impose various development, diligence, commercialization, and other obligations on us. For example, we may be required to use commercially reasonable efforts to engage in various development and commercialization activities with respect to licensed products, and satisfy specified milestone and royalty payment obligations. In spite of our efforts, our licensors might conclude that we have materially breached our obligations under such license agreements and might therefore terminate the license agreements, thereby removing or limiting our ability to develop and commercialize products and technology covered by these license agreements. If these in-licenses are terminated, or if the underlying patents fail to provide the intended exclusivity, competitors or other third parties would have the freedom to seek regulatory approval of, and to market, products identical to ours and we may be required to cease our development and commercialization of product candidates that we may identify. Any of the foregoing could have a material adverse effect on our competitive position, business, financial conditions, results of operations, and prospects. Moreover, disputes may arise regarding intellectual property subject to a licensing agreement, including:
| ● | the scope of rights granted under the license agreement and other interpretation-related issues; | |
| ● | the extent to which our product candidates, technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement; | |
| ● | the sublicensing of patent and other rights under our collaborative development relationships; | |
| ● | our diligence obligations under the license agreement and what activities satisfy those diligence obligations; | |
| ● | the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners; and | |
| ● | the priority of invention of patented technology. In addition, the agreements under which we currently or in the future license intellectual property or technology from third parties are complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology, or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of operations, and prospects. Moreover, if disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on commercially acceptable terms, we may be unable to successfully develop and commercialize the affected product candidates, which could have a material adverse effect on our business, financial conditions, results of operations, and prospects. |
| 44 |
We may infringe the intellectual property rights of others, which may prevent or delay our product development efforts and stop us from commercializing, or increase the costs of commercializing, Aramchol or Amilo-5MER or any other product candidate.
Our commercial success depends significantly on our ability to operate without infringing the patents and other intellectual property rights of third parties. For example, there could be issued patents of which we are not aware that our product candidate infringes. There also could be patents that we believe we do not infringe, but that we may ultimately be found to infringe. Moreover, patent applications are in some cases maintained in secrecy until patents are issued. The publication of discoveries in the scientific or patent literature frequently occurs substantially later than the date on which the underlying discoveries were made and patent applications were filed. Because patents can take many years to issue, there may be currently pending applications of which we are unaware that may later result in issued patents that our product candidates infringe. For example, pending applications may exist that provide support or can be amended to provide support for a claim that results in an issued patent that our product candidates infringes.
Third parties may assert that we are employing their proprietary technology without authorization. If a court held that any third-party patents are valid, enforceable and cover any of our product candidates or their use, the holders of any of these patents may be able to block our ability to commercialize any such product candidate unless we obtained a license under the applicable patents, or until the patents expire. In addition to litigation proceedings which may be filed against us, we may not be able to enter into licensing arrangements or make other arrangements at a reasonable cost or on reasonable terms. Any inability to secure licenses or alternative technology could result in delays in the introduction of our product candidates or lead to prohibition of the manufacture or sale of products by us.
We may become involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive, time consuming and unsuccessful.
Competitors may infringe our patents or other intellectual property. Although we are not currently involved in any litigation, if we were to initiate legal proceedings against a third party to enforce a patent covering our product candidates, the defendant could counterclaim that the patent covering our product candidate is invalid and/or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness, written description or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. The outcome following legal assertions of invalidity and unenforceability is unpredictable.
Interference or derivation proceedings provoked by third parties or brought by us or declared by the USPTO may be necessary to determine the priority of inventions with respect to our patents or patent applications. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms or at all, or if a non-exclusive license is offered and our competitors gain access to the same technology. Our defense of litigation or interference or derivation proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. In addition, the uncertainties associated with litigation could have a material adverse effect on our ability to raise the funds necessary to continue our clinical trials, continue our research programs, license necessary technology from third parties, or enter into development partnerships that would help us bring our product candidates to market.
| 45 |
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions, or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of our ordinary shares.
We may be unable to adequately prevent disclosure and unauthorized use of trade secrets and other proprietary information by third parties.
Our ability to obtain and maintain patent protection and trade secret protection for our intellectual property and proprietary technologies, our product candidates and their uses is important to our commercial success. We rely on a combination of patent, copyright, trademark and trade secret laws, non-disclosure and confidentiality agreements, licenses, assignment of inventions agreements and other restrictions on disclosure and use to protect our intellectual property rights.
We also rely on trade secrets to protect our proprietary know-how and technological advances, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to protect our trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover our trade secrets and proprietary information. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights. Failure to obtain or maintain trade secret protection could enable competitors to use our proprietary information to develop products that compete with our product candidates or cause additional material adverse effects upon our competitive business position.
We cannot be certain that the steps that we have taken will prevent the misappropriation or other violation of our confidential information and other intellectual property, particularly in foreign countries in which laws may not protect our proprietary rights as fully as in the United States and other developed economies. Moreover, if we lose any key personnel, we may not be able to prevent the unauthorized disclosure or use of our technical knowledge or other trade secrets by those former employees. If we are unable to maintain the security of our proprietary technology, this could materially adversely affect our competitive advantage, business and results of operations.
Under applicable U.S. and Israeli law, we may not be able to enforce covenants not to compete and therefore may be unable to prevent our competitors from benefiting from the expertise of some of our former employees. In addition, employees may be entitled to seek compensation for their inventions irrespective of their agreements with us, which in turn could impact our future profitability.
We generally enter into non-competition agreements with our employees and certain key consultants, or our employment and consulting agreements contain non-competition provisions. These agreements, to the extent they are in place and in effect, prohibit our employees and certain key consultants, if they cease working for us, from competing directly with us or working for our competitors or clients for a limited period of time. We may be unable to enforce these agreements under the laws of the jurisdictions in which our employees work and it may be difficult for us to restrict our competitors from benefitting from the expertise our former employees or consultants developed while working for us. For example, Israeli courts have required employers seeking to enforce non-compete undertakings of a former employee to demonstrate that the competitive activities of the former employee will harm one of a limited number of material interests of the employer which have been recognized by the courts, such as the secrecy of a company’s confidential commercial information or the protection of its intellectual property. If we cannot demonstrate that such interests will be harmed, we may be unable to prevent our competitors from benefiting from the expertise of our former employees or consultants and our ability to remain competitive may be diminished.
| 46 |
In addition, under the Israeli Patent Law, 5727-1967, or the Patent Law, inventions conceived by an employee in the course and as a result of or arising from his or her employment with a company are regarded as “service inventions,” which belong to the employer, absent a specific agreement between the employee and employer giving the employee service invention rights. The Patent Law also provides that if there is no such agreement between an employer and an employee, the Israeli Compensation and Royalties Committee, or the Committee, a body constituted under the Patent Law, shall determine whether the employee is entitled to remuneration for his or her inventions. Case law clarifies that the right to receive consideration for “service inventions” can be waived by the employee and that in certain circumstances, such waiver does not necessarily have to be explicit. The Committee will examine, on a case-by-case basis, the general contractual framework between the parties, using interpretation rules of the general Israeli contract laws. Further, the Committee has not yet determined one specific formula for calculating this remuneration, but rather uses the criteria specified in the Patent Law. Although we generally enter into assignment-of-invention agreements with our employees pursuant to which such individuals assign to us all rights to any inventions created in the scope of their employment or engagement with us, we may face claims demanding remuneration in consideration for assigned inventions. As a consequence of such claims, we could be required to pay additional remuneration or royalties to our current and/or former employees, or be forced to litigate such claims, which could negatively affect our business.
Any lawsuits relating to infringement of intellectual property rights necessary to defend ourselves or enforce our rights will be costly and time consuming.
We may be required to initiate litigation to enforce our rights or defend our activities in response to alleged infringement of a third-party. In addition, we may be sued by others who hold intellectual property rights and who claim that their rights are infringed by our product candidates. These lawsuits can be very time consuming and costly. There is a substantial amount of litigation involving patent and other intellectual property rights in the biotechnology and pharmaceutical industries generally.
A third-party may claim that we are using inventions claimed by their patents and may go to court to stop us from engaging in our normal operations and activities, such as research, development, and the sale of any future products. Such lawsuits are expensive and would consume time and other resources. There is a risk that such court will decide that we are infringing the third-party’s patents and will order us to stop the activities claimed by the patents, redesign our products or processes to avoid infringement or obtain licenses, which may not be available on commercially reasonable terms. In addition, there is a risk that a court will order us to pay the other party damages for infringement.
Moreover, there is no guarantee that any prevailing patent owner would offer us a license so that we could continue to engage in activities claimed by the patent, or that such a license, if made available to us, could be acquired on commercially acceptable terms. In addition, third parties may, in the future, assert other intellectual property infringement claims against us with respect to other product candidates, technologies or other matters.
In addition, our patents and patent applications could face challenges. Any of these challenges, if successful, could result in the invalidation of, or in a narrowing of the scope of, any of our patents and patent applications subject to challenge. Any of these challenges, regardless of their success, would likely be time consuming and expensive to defend and resolve and would divert our management’s time and attention.
| 47 |
Changes in patent law could diminish the value of patents in general, thereby impairing our ability to protect Aramchol, Amilo-5MER or any other product candidate.
As is the case with other pharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involve both technological and legal complexity. Therefore, obtaining and enforcing pharmaceutical patents is costly, time-consuming, and inherently uncertain. In particular, the United States has recently enacted, and is currently implementing, wide-ranging patent reform legislation. The United States Supreme Court has ruled on several patent cases in recent years, and could do so again in the future, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty regarding our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by applicable courts and legislatures in the countries in which we may pursue patent protection, including those of the U.S. Congress, the federal courts and the USPTO, the laws and regulations governing patents and the interpretations of such laws could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
Obtaining and maintaining our patent protection depends on compliance with various procedural, documentary, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case.
Risks Related to Ownership of Our Ordinary Shares
We are currently operating in a period of economic uncertainty and capital markets disruption, which has been significantly impacted by geopolitical instability due to the ongoing military conflict between Russia and Ukraine.
U.S. and global markets are experiencing volatility and disruption following the escalation of geopolitical tensions and the start of the military conflict between Russia and Ukraine. In February 2022, Russia launched a full-scale military invasion of Ukraine. Although the length and impact of the ongoing military conflict is highly unpredictable, the conflict in Ukraine could lead to market disruptions, including significant volatility in commodity prices, credit and capital markets. Additionally, Russia’s prior annexation of Crimea, recent recognition of two separatist republics in the Donetsk and Luhansk regions of Ukraine and subsequent military interventions in Ukraine have led to sanctions and other penalties being levied by the United States, European Union and other countries against Russia, Belarus, the Crimea Region of Ukraine, the so-called Donetsk People’s Republic, and the so-called Luhansk People’s Republic, including agreement to remove certain Russian financial institutions from the Society for Worldwide Interbank Financial Telecommunication (SWIFT) payment system. Additional potential sanctions and penalties have also been proposed and/or threatened. Russian military actions and the resulting sanctions could adversely affect the global economy and financial markets and lead to instability and lack of liquidity in capital markets, potentially making it more difficult for us to obtain additional funds. Any of the abovementioned factors could affect our business, prospects, financial condition, and operating results. The extent and duration of the military action, sanctions and resulting market disruptions are impossible to predict, but could be substantial. Any such disruptions may also magnify the impact of other risks described in this Annual Report on Form 20-F.
The market price of our ordinary shares is volatile and you may sustain a complete loss of your investment.
Since our initial public offering, the trading price of our ordinary shares has been volatile and is likely to continue to be volatile. In addition, the trading volume is and has been volatile and oftentimes relatively illiquid. The following factors, some of which are beyond our control, in addition to other risk factors described in this section, may have a significant impact on the market price and trading volume of our ordinary shares:
| ● | delays in existing clinical trials; | |
| ● | inability to obtain the approvals necessary to commence further clinical trials; |
| 48 |
| ● | unsatisfactory or inconclusive results of clinical trials; | |
| ● | termination of clinical trials; | |
| ● | adverse events in our ongoing clinical trials; | |
| ● | announcements of regulatory approval or the failure to obtain it, or specific label indications or patient populations for its use, or changes or delays in the regulatory review process; | |
| ● | announcements of therapeutic innovations or new products by us or our competitors; | |
| ● | adverse actions taken by regulatory agencies with respect to our clinical trials, manufacturing supply chain or sales and marketing activities; | |
| ● | changes or developments in laws or regulations applicable to our product candidates; | |
| ● | any adverse changes to our relationship with manufacturers or suppliers; | |
| ● | any product liability actions or intellectual property infringement actions in which we may become involved; | |
| ● | announcements concerning our competitors or the pharmaceutical industry in general; | |
| ● | achievement of expected product sales and profitability or our failure to meet expectations; | |
| ● | our commencement of, or involvement in, litigation; | |
| ● | any major changes in our board of directors, management or other key personnel; | |
| ● | legislation in the United States, Europe and other foreign countries relating to the sale or pricing of pharmaceuticals; | |
| ● | announcements by us of significant strategic partnerships, out-licensing, in-licensing, joint ventures, acquisitions or capital commitments; | |
| ● | expiration or terminations of licenses, research contracts or other collaboration agreements; | |
| ● | public concern as to the safety of drugs we, our licensees or others develop; | |
| ● | success of research and development projects; | |
| ● | variations in our and our competitors’ results of operations; | |
| ● | changes in earnings estimates, cash flow guidance, or recommendations by securities analysts; | |
| ● | developments by our licensees, if any; | |
| ● | future issuances of ordinary shares or other securities; and | |
| ● | natural disasters and political and economic instability, including wars, terrorism, political unrest, results of certain elections and votes, emergence of a pandemic, or other widespread health emergencies (or concerns over the possibility of such an emergency, including for example, the COVID-19 pandemic), boycotts, adoption or expansion of government trade restrictions, and other business restrictions. |
| 49 |
These factors and any corresponding price fluctuations may materially and adversely affect the market price and trading volume of our ordinary shares and result in substantial losses by our investors.
In addition, the stock market in general, and the Nasdaq Capital Market and the market for biotechnology companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of our Company and that of small companies. Broad market and industry factors may negatively affect the market price of our ordinary shares, regardless of our actual operating performance. Further, a systemic decline in the financial markets and related factors beyond our control may cause our share price to decline rapidly and unexpectedly. Price volatility of our ordinary shares might be worse if the trading volume of our ordinary shares is low. Following periods of market volatility or a material decrease in the value of our ordinary shares, shareholders may institute securities class action litigation. If we were involved in securities litigation, it could have a substantial cost and divert resources and attention of management from our business, even if we are successful. Future sales of our ordinary shares could also reduce the market price of such stock. Any adverse determination in litigation could also subject us to significant liabilities.
Moreover, the liquidity of our ordinary shares has been limited, not only in terms of the number of shares that can be bought and sold at a given price, but by delays in the timing of transactions and reduction in security analysts’ and the media’s coverage of us, if any. These factors may result in lower prices for our ordinary shares than might otherwise be obtained and could also result in a larger spread between the bid and ask prices for our ordinary shares. In addition, without a large float, our ordinary shares are less liquid than the stock of companies with broader public ownership and, as a result, the trading prices of our ordinary shares are more volatile. In the absence of an active public trading market, an investor may be unable to liquidate its investment in our ordinary shares. Trading of a relatively small volume of our ordinary shares may have a greater impact on the trading price of our stock than would be the case if our public float were larger. We cannot predict the prices at which our ordinary shares will trade in the future.
Our ordinary shares are listed on the Nasdaq Capital Market. As such, we must meet the Nasdaq Capital Market’s continued listing requirements and other Nasdaq rules, or we may risk delisting. Delisting could negatively affect the price of our ordinary shares, which could make it more difficult for us to sell securities in a financing and for you to sell your ordinary shares.
Our ordinary shares are listed on the Nasdaq Capital Market. As such, we are required to meet the continued listing requirements of the Nasdaq Capital Market and other Nasdaq rules, including those regarding director independence and independent committee requirements, minimum shareholders’ equity, minimum share price and certain other corporate governance requirements. In particular, we are required to maintain a minimum bid price for our listed ordinary shares of $1.00 per share. If we do not meet these continued listing requirements, our ordinary shares could be delisted. Delisting of our ordinary shares from the Nasdaq Capital Market would cause us to pursue eligibility for trading on other markets or exchanges, or on the pink sheets. In such case, our shareholders’ ability to trade, or obtain quotations of the market value of, our ordinary shares would be severely limited because of lower trading volumes and transaction delays. These factors could contribute to lower prices and larger spreads in the bid and ask prices for our securities. There can be no assurance that our ordinary shares, if delisted from the Nasdaq Capital Market in the future, would be listed on a national securities exchange, a national quotation service, the Over-The-Counter Markets or the pink sheets. Delisting from the Nasdaq Capital Market, or even the issuance of a notice of potential delisting, would also result in negative publicity, make it more difficult for us to raise additional capital, adversely affect the market liquidity of our ordinary shares, reduce security analysts’ coverage of us and diminish investor, supplier and employee confidence. Additionally, the threat of delisting or a delisting of our ordinary shares from the Nasdaq Capital Market, could reduce the number of investors willing to hold or acquire our ordinary shares, thereby further restricting our ability to obtain equity financing, and it could reduce our ability to retain, attract and motivate our directors, officers and employees. In addition, as a consequence of any such delisting, our share price could be negatively affected and our shareholders would likely find it more difficult to sell, or to obtain accurate quotations as to the prices of, our ordinary shares.
| 50 |
Our President and Chief Executive Officer beneficially owns approximately 17.1% of our outstanding ordinary shares, as of April 15, 2022. Therefore, our principal shareholders will be able to exert significant control over matters submitted to our shareholders for approval.
Our President and Chief Executive Officer currently beneficially owns approximately 17.1% of our outstanding ordinary shares as of April 15, 2022. Therefore, our President and Chief Executive Officer will be able to exert significant control over matters submitted to our shareholders for approval. As our President and Chief Executive Officer could significantly influence or even unilaterally approve matters requiring approval by our shareholders, including the election of directors and the approval of mergers or other business combination transactions. The interests of our President and Chief Executive Officer may not always coincide with our interests or the interests of other shareholders. This significant concentration of share ownership may adversely affect the trading price for our ordinary shares because investors often perceive disadvantages in owning stock in companies with controlling shareholders.
Sales of a substantial number of our ordinary shares in the public market could cause our share price to fall.
Sales of a substantial number of our ordinary shares in the public market, or the perception that these sales might occur, could depress the market price of our ordinary shares and could impair our ability to raise capital through the sale of additional equity securities. We are unable to predict the effect that sales may have on the prevailing market price of our ordinary shares. To date, the lock-up period has expired and substantially all of our outstanding shares are eligible for unrestricted sale. Sales of shares by these shareholders would likely result in the supply of our ordinary shares far exceeding the demand for our ordinary shares and could have a material adverse effect on the trading price of our ordinary shares.
Raising additional capital would cause dilution to our existing shareholders, and may restrict our operations or require us to relinquish rights.
We may seek additional capital through a combination of private and public equity offerings, “at-the-market” issuances, equity-linked and structured transactions, debt (straight, convertible, or otherwise) financings, collaborations and licensing arrangements. Under our existing “at the market” equity offering program as of April 28, 2022, we may sell, from time to time, up to approximately $50.0 million of additional ordinary shares subject to limitations under the Baby Shelf Rule. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms may include liquidation or other preferences that adversely affect your rights as a shareholder. Debt financing, if available, would result in increased fixed payment obligations and may involve agreements that include covenants limiting or restricting our ability to take specific actions such as incurring debt, making capital expenditures or declaring dividends. If we raise additional funds through collaboration, strategic alliance and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams or product candidates, or grant licenses on terms that are not favorable to us. Depending upon market liquidity at the time, additional sales of shares registered at any given time could cause the trading price of our ordinary shares to decline.
| 51 |
Our U.S. shareholders may suffer adverse tax consequences due to our expected classification as a passive foreign investment company.
Generally, if for any taxable year 75% or more of our gross income is passive income, or at least 50% of the average value of our assets is attributable to assets that are held for the production of, or produce, passive income, we would be characterized as a passive foreign investment company, or PFIC, for U.S. federal income tax purposes. Based upon our review of our financial data, we believe that we were a PFIC for our 2021 taxable year and expect to be a PFIC for the 2022 taxable year. Because PFIC status is determined annually and is based on our income, assets and activities for the entire taxable year, it is not possible to determine with certainty whether we will be characterized as a PFIC for the 2022 taxable year until after the close of the year, and there can be no assurance that we will not be classified as a PFIC in any future year. If we were to be characterized as a PFIC for U.S. federal income tax purposes in any taxable year during which a U.S. Holder (as defined below) owns ordinary shares, such U.S. Holder could face adverse U.S. federal income tax consequences. For example, such U.S. Holder could be subject to additional taxes and interest charges upon certain distributions by us and any gain recognized on a sale, exchange or other disposition of our shares, whether or not we continue to be characterized as a PFIC. Certain adverse consequences of PFIC status can be mitigated if a U.S. Holder makes a “mark to market” election or an election to treat us as a qualified electing fund, or QEF. Upon request, we expect to provide the information necessary for U.S. Holders to make “qualified electing fund elections” if we are classified as a PFIC. Each investor is urged to consult its tax advisor with respect to the application of the PFIC rules. See also “Item 10. Additional Information—E. Taxation— Certain U.S. Federal Income Tax Considerations.”
If the securities analysts that currently cover our stock, or will do so in the future, or industry analysts do not publish or cease publishing research or reports about us, our business or our market, or if they adversely change their recommendations or publish negative reports regarding our business or our shares, our share price and trading volume could be negatively impacted.
The trading market for our ordinary shares is influenced by the research and reports that industry or securities analysts may publish about us, our business, our market or our competitors. We do not have any control over these analysts and we cannot provide any assurance that analysts will cover us or provide favorable coverage. If any of the analysts who do cover, or may cover us in the future, adversely change their recommendation regarding our shares, or provide more favorable relative recommendations about our competitors, our share price would likely decline. If any analyst who cover us cease coverage of our company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could negatively impact our share price or trading volume.
Because we do not intend to declare cash dividends on our ordinary shares in the foreseeable future, shareholders must rely on appreciation of the value of our ordinary shares for any return on their investment.
We have never declared or paid cash dividends on our ordinary shares. We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends in the foreseeable future. Moreover, the Israeli Companies Law, 5759-1999, or the Companies Law, imposes certain restrictions on our ability to declare and pay dividends. See “Item 8. Financial Information—Consolidated Financial Statements and Other Financial Information—Dividend Policy” for additional information.
The requirements associated with being a public company require significant company resources and management attention.
We are subject to the reporting requirements of the Securities Exchange Act of 1934, or the Exchange Act, the Sarbanes-Oxley Act, the listing requirements of the Nasdaq Capital Market, on which our ordinary shares are traded, and other applicable securities rules and regulations. The Exchange Act requires that we file periodic reports with respect to our business and financial condition and maintain effective disclosure controls and procedures and internal control over financial reporting. In addition, subsequent rules implemented by the SEC and the Nasdaq Capital Market may also impose various additional requirements on public companies. As a result, we incurred and will continue to incur additional legal, accounting and other expenses that we did not incur as a privately-held company, particularly since, as of December 31, 2020, we are no longer considered an “emerging growth company” as defined in the JOBS Act. Further, the need to establish the corporate infrastructure demanded of a public company may divert management’s attention from implementing our development plans. We have made and will continue to make changes to our corporate governance standards, compensation policy, disclosure controls and financial reporting and accounting systems to meet our reporting obligations and applicable law. The measures we take, however, may not be sufficient to satisfy our obligations as a public company, which could subject us to delisting of our ordinary shares, fines, sanctions and other regulatory action and potentially civil litigation.
| 52 |
As a “foreign private issuer,” we are permitted to and currently do follow certain home country corporate governance practices instead of otherwise applicable SEC and Nasdaq Capital Market requirements, which may result in less protection than is accorded to investors under rules applicable to domestic U.S. issuers.
As a “foreign private issuer,” we are permitted to, and currently do, follow certain home country corporate governance practices instead of those otherwise required under the Listing Rules of the Nasdaq Capital Market, or the Nasdaq Listing Rules, for domestic U.S. issuers. For instance, we currently follow home country practice in Israel with regard to, among other things, director nomination procedure and approval of compensation of officers. In addition, we may follow our home country law instead of the Nasdaq Listing Rules that require that we obtain shareholder approval for certain dilutive events, such as the establishment or amendment of certain equity based compensation plans, an issuance that will result in a change of control of the company, certain transactions other than a public offering involving issuances of a 20% or greater interest in the company, and certain acquisitions of the stock or assets of another company. Following our home country governance practices as opposed to the requirements that would otherwise apply to a U.S. company listed on the Nasdaq Capital Market may provide less protection to you than what is accorded to investors under the Nasdaq Listing Rules applicable to domestic U.S. issuers. See “Item 16G. Corporate Governance.”
In addition, as a “foreign private issuer,” we are exempt from the rules and regulations under the Exchange Act related to the furnishing and content of proxy statements and certain individual executive compensation information, and our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. Furthermore, foreign private issuers are not required to file their annual report on Form 20-F until 120 days after the end of each fiscal year, while U.S. domestic issuers that are accelerated filers are required to file their annual report on Form 10-K within 75 days after the end of each fiscal year and U.S. domestic issuers that are large accelerated filers are required to file their annual report on Form 10-K within 60 days after the end of each fiscal year. Additionally, as a “foreign private issuer,” we are also not subject to the requirements of Regulation FD (Fair Disclosure) promulgated under the Exchange Act. These exemptions and leniencies reduce the frequency and scope of information and protections to which you are entitled as an investor.
If our ordinary shares become a “penny stock,” it may be more difficult for investors to sell their ordinary shares, and the market price of our ordinary shares may be adversely affected.
Our ordinary shares could become a “penny stock” if, among other things, the share price is below $5.00 per share, we are not listed on a national securities exchange or we have not met certain net tangible asset or average revenue requirements. Broker-dealers who sell penny stocks must provide purchasers of these stocks with a standardized risk-disclosure document prepared by the SEC. This document provides information about penny stocks and the nature and level of risks involved in investing in the penny-stock market. A broker must also give a purchaser, orally or in writing, bid and offer quotations and information regarding broker and salesperson compensation, make a written determination that the penny stock is a suitable investment for the purchaser, and obtain the purchaser’s written agreement to the purchase. Broker-dealers must also provide customers that hold penny stock in their accounts with such broker-dealer a monthly statement containing price and market information relating to the penny stock. If a penny stock is sold to an investor in violation of the penny stock rules, the investor may be able to cancel its purchase and get its money back.
If applicable, the penny stock rules may make it difficult for investors to sell their ordinary shares. Because of the rules and restrictions applicable to a penny stock, there is less trading in penny stocks and the market price of our ordinary shares may be adversely affected. Also, many brokers choose not to participate in penny stock transactions. Accordingly, investors may not always be able to resell their ordinary shares publicly at times and prices that they feel are appropriate and the market price of our ordinary shares may be adversely affected.
| 53 |
Risks Related to Israeli Law and Our Operations in Israel
Our headquarters and other significant operations are located in Israel and, therefore, our results may be adversely affected by political, economic and military instability in Israel.
Our executive offices are located in Tel Aviv, Israel. In addition, the majority of our officers and directors are residents of Israel. Accordingly, political, economic and military conditions in Israel may directly affect our business. Since the establishment of the State of Israel in 1948, a number of armed conflicts have taken place between Israel and its neighboring countries. Any hostilities involving Israel or the interruption or curtailment of trade between Israel and its trading partners could adversely affect our operations and results of operations. In recent years, these have included hostilities between Israel and Hezbollah in Lebanon and Hamas in the Gaza strip, both of which resulted in rockets being fired into Israel, causing casualties and disruption of economic activities. In addition, Israel faces threats from more distant neighbors, in particular, Iran.
Since February 2011, riots and uprisings in several countries in the Middle East and neighboring regions have led to severe political instability in several neighboring states and to a decline in the regional security situation. Such instability may affect the local and global economy, could negatively affect business conditions and, therefore, could adversely affect our operations. To date, these matters have not had any material effect on our business and results of operations; however, the regional security situation and worldwide perceptions of it are outside our control, and there can be no assurance that these matters will not negatively affect us in the future. In addition, the political and security situation in Israel may result in parties with whom we have agreements involving performance in Israel claiming that they are not obligated to perform their commitments under those agreements pursuant to force majeure provisions in such agreements.
Our commercial insurance does not cover losses that may occur as a result of an event associated with the security situation in the Middle East. Although the Israeli government is currently committed to covering the reinstatement value of direct damages that are caused by terrorist attacks or acts of war, we cannot assure you that this government coverage will be maintained, or if maintained, will be sufficient to compensate us fully for damages incurred. Any losses or damages incurred by us could have a material adverse effect on our business. Any armed conflicts or political instability in the region would likely negatively affect business conditions generally and could harm our results of operations.
Further, in the past, the State of Israel and Israeli companies have been subjects of economic boycotts. Several countries still restrict business with the State of Israel and with Israeli companies. These restrictive laws and policies may have an adverse impact on our operating results, financial condition or the expansion of our business.
Our operations may be disrupted as a result of the obligation of Israeli citizens to perform military service.
Many Israeli citizens are obligated to perform several days, and in some cases more, of annual military reserve duty until they reach the age of 40 (or older, for reservists who are officers or who have certain occupations) and, in the event of a military conflict, may be called to active duty. In response to increases in terrorist activity, there have been periods of significant call-ups of military reservists. It is possible that there will be military reserve duty call-ups in the future. Our operations could be disrupted by such call- ups, which may include the call-up of our employees or the employees of our Israeli business partners. Such disruption could materially adversely affect our business, financial condition and results of operations.
| 54 |
Exchange rate fluctuations between the U.S. dollar and the New Israeli Shekel currencies may negatively affect our earnings.
Our functional currency is the U.S. dollar. We incur expenses in U.S. dollars and New Israeli Shekels, or NIS. As a result, we are exposed to the risks that the NIS may appreciate relative to the U.S. dollar, or, if either the NIS devalues relative to the U.S. dollar, that the inflation rate in Israel may exceed such rate of devaluation of the NIS, or that the timing of such devaluation may lag behind inflation in Israel. In any such event, the U.S. dollar cost of our operations in Israel would increase and our U.S. dollar-denominated results of operations would be adversely affected. The average exchange rate for the year ended December 31, 2021 was $1.00 = NIS 3.110. We cannot predict any future trends in the rate of inflation in Israel or the rate of devaluation, if any, of the NIS against the U.S. dollar. As of the date hereof, neither the inflation rate in Israel has exceeded the rate of devaluation of the NIS, respectively, during the calendar years 2019, 2020 or 2021.
Provisions of Israeli law and our articles of association, or Articles, may delay, prevent or otherwise impede a merger with, or an acquisition of, our company, which could prevent a change of control, even when the terms of such a transaction are favorable to us and our shareholders.
The Companies Law regulates, among others, mergers, requires tender offers for acquisitions of shares above specified thresholds, requires special approvals for transactions involving directors, officers or significant shareholders and regulates other matters that may be relevant to such types of transactions. See “Item 10. Additional Information—B. —Mergers and Acquisitions under Israeli Law” for additional information.
Furthermore, Israeli tax considerations may make potential transactions unappealing to us or to our shareholders whose country of residence does not have a tax treaty with Israel exempting such shareholders from Israeli tax. See “Item 10. Additional Information—E. Taxation—Certain Israeli Tax Considerations” for additional information.
Moreover, the classification of our Board into three classes with terms of approximately three years each, per our Articles, the requirement of affirmative vote of at least 75% of the voting rights of the Company represented personally or by proxy and voting thereon at a general meeting in order to amend or replace our Articles, together with the other provisions of the Articles and Israeli law, could deter or delay potential future merger, acquisition, tender or takeover offers, proxy contests or changes in control or management of the Company.
It may be difficult to enforce a judgment of a United States court against us, our officers, directors and the Israeli experts named in this annual report in Israel or the United States, to assert United States securities laws claims in Israel or to serve process on our officers, directors and these experts.
We were and continue to be organized in Israel. Most of our executive officers and directors reside outside of the United States, and all of our assets and most of the assets of these persons are located outside of the United States. Therefore, a judgment obtained against us, or any of these persons, including a judgment based on the civil liability provisions of the U.S. federal securities laws, may not be collectible in the United States and may not necessarily be enforced by an Israeli court. It also may be difficult to effect service of process on these persons in the United States or to assert U.S. securities law claims in original actions instituted in Israel. Additionally, it may be difficult for an investor, or any other person or entity, to initiate an action with respect to United States securities laws in Israel. Israeli courts may refuse to hear a claim based on an alleged violation of United States securities laws reasoning that Israel is not the most appropriate forum in which to bring such a claim. In addition, even if an Israeli court agrees to hear a claim, it may determine that Israeli law and not United States law is applicable to the claim. If United States law is found to be applicable, the content of applicable United States law must be proven as a fact by expert witnesses, which can be a time consuming and costly process. Certain matters of procedure will also be governed by Israeli law. There is little binding case law in Israel that addresses the matters described above. As a result of the difficulty associated with enforcing a judgment against us in Israel, our shareholders may not be able to collect any damages awarded by either a United States or foreign court.
| 55 |
Your rights, liabilities and responsibilities as a shareholder will be governed by Israeli law and differ in some material respects from those under U.S. law.
Because we are an Israeli company, the rights and responsibilities of our shareholders are governed by our Articles and Israeli law. These rights, liabilities and responsibilities differ in some material respects from the rights, liabilities and responsibilities of shareholders in a U.S. corporation. In particular, a shareholder of an Israeli company has a duty to act in good faith towards the company and other shareholders and to refrain from abusing his, her or its power in the company, including, among other things, when voting at the general meeting of shareholders on certain matters. Israeli law provides that these duties are applicable to shareholder votes on, among other things, amendments to a company’s articles of association, increases in a company’s authorized share capital, mergers and interested party transactions requiring shareholder approval. In addition, a controlling shareholder, a shareholder who knows that it possesses the power to determine the outcome of a shareholders’ vote or a shareholder who has the power to appoint or prevent the appointment of a director or executive officer in the company, has a duty of fairness towards the company. However, Israeli law does not define the substance of this duty of fairness. There is little case law available to assist in understanding the implications of these provisions that govern shareholder behavior. These provisions may be interpreted to impose additional obligations and liabilities on holders of our ordinary shares that are not typically imposed on shareholders of U.S. corporations. See “Item 10. Additional Information—B Memorandum and Articles of Association—Shareholder Duties” for additional information.
Any of the risk factors referred to above could significantly and negatively affect our business, results of operations or financial condition, which may reduce our ability to pay dividends and lower the trading price of our ordinary shares. The risks referred to above are not the only ones that may exist. Additional risks not currently known by us or that we deem immaterial may also impair our business operations.
ITEM 4. Information on the Company.
A. Historical Background and Corporate Structure
Galmed Pharmaceuticals Ltd., was incorporated in Israel on July 31, 2013 as a privately held company and is governed by the Companies Law. However, our business has been operating since 2000 under a different group of companies established in the same year, or the Group. Originally, we operated under the parent company, GHI. GHI held all of the equity rights in and to Galmed 2000 Inc., a holdings company incorporated in the British Virgin Islands, or GTTI. GTTI held all of the equity rights in and to Galmed International Limited, a company incorporated in Malta, or GIL (other than 0.1% of the share capital held by GHI). GIL held all of the equity rights in and to Galmed Medical Research Ltd., an Israeli company, or GMR. Our intellectual property was held by GIL. The research and development was conducted by GMR as a service to GIL on a cost plus basis. GIL was responsible for all product development.
On February 2, 2014, we underwent the Reorganization, pursuant to which all of our intangible assets (including our intellectual property) were transferred from GIL to Galmed Research and Development Ltd., or GRD. The Reorganization was effectuated by share transfers and asset transfers, resulting in the Company as the parent company and 100% equity-owner of the following companies: (1) GRD, which holds all the Group’s intellectual property, including the Company’s patent portfolio; (2) GIL, which is an inactive company; and (3) GTTI, which was liquidated in 2017. GIL held GMR, which became an inactive company in 2015 and was liquidated in February 2019. The Reorganization was conducted in order to simplify our capital structure, reduce our operating cost and to improve our ability to raise funds. Immediately prior to the Reorganization, all our shareholders collectively held 9,739 ordinary shares of GHI. In connection with the Reorganization, and in accordance with the Tax Pre-Ruling, we issued to all such shareholders ordinary shares of the Company, such that upon the Reorganization all our shareholders collectively held 7,099,731 ordinary shares of the Company, in the same proportion among all shareholders, which reflected a ratio of 729 ordinary shares of the Company for each ordinary share of GHI.
| 56 |
The following is a diagram of our corporate structure (following GTTI’s liquidation):
On March 18, 2014, we completed our initial public offering and since then have been listed on the Nasdaq Capital Market under the symbol “GLMD”.
Our principal executive offices and registered office in Israel are located at 16 Tiomkin Street, Tel Aviv, Israel, 6578317 and our telephone number is +972-3-693-8448. Our website address is http://www.galmedpharma.com. The information contained on, or that can be accessed through, our website is neither a part of nor incorporated into this annual report. We have included our website address in this annual report solely as an inactive textual reference. Puglisi & Associates, or Puglisi, serves as our authorized representative in the United States for certain limited matters. Puglisi’s address is 850 Library Avenue, Newark, Delaware 19711.
The SEC maintains an internet site that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC at http://sec.gov. We use our website (http://www.galmedpharma.com) as a channel of distribution of Company information. The information we post through this channel may be deemed material. Accordingly, investors should monitor our website, in addition to following our press releases, SEC filings and public conference calls and webcasts. The contents of our website are not, however, a part of this annual report.
Other than as described in “Item 5. Operating and Financial Review and Prospects—Contractual Obligations”, we have not had any material commitments for capital expenditures, including any anticipated material acquisition of plant and equipment or interests in other companies, since January 1, 2014. Additionally, we have not had any material capital divestitures since January 1, 2014.
B. Business Overview
We are a clinical-stage biopharmaceutical company focused on the development of Aramchol, a liver targeted stearoyl-coenzyme A desaturase-1, or SCD1, modulator, first in class, novel, oral therapy for the treatment of NASH for variable populations. We are also collaborating with the Hebrew University in the development of Amilo-5MER, a 5 amino acid synthetic peptide.
We believe that our lead product candidate, Aramchol, has the potential to be a disease modifying treatment for fatty liver disorders, including NASH, which is a chronic disease that constitutes a large unmet medical need.
Aramchol is a synthetic conjugate of cholic acid, or a type of bile acid, and arachidic acid, or a type of saturated fatty acid, both of which, in their non-synthetic forms, are naturally occurring. The conjugated molecule acts upon important metabolic pathways, reducing fat accumulation in the liver, improving fatty acid oxidation and regulating the transport of cholesterol. The ability of Aramchol to decrease liver fat content may also reduce the inflammation and fibrosis in the liver and the risk of cardiovascular complications associated with NASH. Pre-clinical studies suggest Aramchol’s effect on fibrosis is also direct via collagen production from human hepatic stellate cells. We believe that Aramchol’s ability to reduce liver fat and liver fibrosis and the safety profile observed to date will enable it to be a treatment for all stages of NASH in patients who are overweight or obese and have pre diabetes or type II diabetes mellitus and prevent the hepatic complications associated therewith.
| 57 |
In September 2019, we initiated our Phase 3 ARMOR Study to evaluate the efficacy and safety of Aramchol in subjects with NASH and fibrosis. The ARMOR Study was originally comprised of two parts, a randomized, double-blind, placebo-controlled histology-based registrational part and a clinically based part where subjects will continue with the same treatment for approximately five years. In December 2020, we announced the addition of a 150-patient open label part to the ARMOR Study and suspended randomization of new patients into the double-blind, placebo-controlled histology-based registrational part of ARMOR as currently enrolled patients are transitioned to the open label part. We are seeking to initiate the double-blind placebo-controlled histology-based registrational part of ARMOR in the second half of 2023, subject to, among other things, the results of our open label part, sufficient funding and clarification of the regulatory approval process for NASH drugs.
In August 2021, we announced that the FDA agreed with our plan to use Aramchol meglumine (in lieu of Aramchol free acid) in our ARMOR Study without the need to conduct additional nonclinical and clinical studies other than planned limited pharmacology studies relating to Aramchol meglumine. In addition, the Medicines and Healthcare products Regulatory Agency, or MHRA, the pharmaceuticals regulator in the UK, also agreed with our plan to proceed with our proposed clinical studies with Aramchol meglumine in lieu of Aramchol free acid without the need to repeat nonclinical and clinical studies other than planned limited pharmacology studies relating to Aramchol meglumine.
In November 2021, we announced positive interim data on the open label part of the ARMOR Study showing clinically significant effect on fibrosis improvement based on histology in the first 16 patients. Subsequently in November 2021, we announced that new analyses of biomarkers corroborate this effect showing statistically significant reductions in biomarkers associated with liver fibrosis including ALT, AST, Fib-4 and ProC-3. Reductions of a similar magnitude were seen in a cohort of the first 20 patients for which paired biopsy have been analyzed and a cohort of 50 patients for which biomarker data was analyzed based on all available data at that time. In addition, we reported that Aramchol continues to show excellent safety and tolerability profile. Data support that a higher dose of Aramchol could provide statistically and clinically meaningful effect on fibrosis in the double-blind placebo controlled part for submission of the ARMOR Study to support an NDA under Sub-part H.
In April 2022, we announced further positive interim data on the open label part of the ARMOR Study showing robust fibrosis improvement across multimodality histological assessment. Results of post baseline biopsies performed either at 24 weeks or at 48 weeks from 46 subjects with NASH and F1-3 that received Aramchol support the anti-fibrotic effect of Aramchol and reinforce the favorable safety profile of Aramchol.
We are also developing Amilo-5MER in a research collaboration between us and the Hebrew University of Jerusalem. In March 2021, we announced the treatment of the first subject in the first in human Phase 1 clinical trial evaluating Amilo-5MER for the treatment of chronic inflammatory diseases and in January 2022, we announced results of the Phase 1 clinical trial of Amilo-5MER in healthy volunteers that demonstrated an excellent safety profile and tolerability.
Non-Alcoholic Fatty Liver Disease (NAFLD) / Non-Alcoholic Steato-Hepatitis (NASH)
It is estimated that the global prevalence of NAFLD, the precondition to NASH, is approximately 25% in the general population and much higher in certain high risk groups. This disease is also now recognized as one of the most common liver disorders, and a significant growing public health problem. In the US alone, 80 - 100 million people are said to be affected by NAFLD, and its prevalence is rapidly growing in parallel with metabolic syndromes, particularly obesity and diabetes.
| 58 |
NAFLD is characterized by the accumulation of fat of 5% or greater in the liver of people who drink alcohol only in moderation, or not at all. There may be numerous causes of NAFLD, however, the disease is mostly associated with a high fat, fructose-rich diet. Although NAFLD is generally asymptomatic, it is a major risk factor for liver inflammation (NASH) and scarring (fibrosis and cirrhosis). In addition, NAFLD is also associated with metabolic syndrome and cardiovascular disease. Currently, NAFLD can only be managed through lifestyle improvements, such as weight reduction and physical activity.
NASH is an emerging world crisis impacting an estimated 3% to 5% of the U.S. population and an estimated 2% to 4% globally, and is associated with increased risk of liver cirrhosis, liver failure, hepatocellular cancer, as well as metabolic and cardiovascular diseases. The major characteristics of NASH are elevated liver fat, inflammation, ballooning and fibrosis.
However, despite the growing need, there are currently no approved therapeutic treatments for NASH. Modification of risk factors, such as obesity and hyperlipidemia, and proper diabetic control is generally recommended for the treatment of NASH, and the standard of care includes lifestyle changes to promote weight loss, including low-calorie, low-fat diets and physical activity. Although weight loss can be potentially significant in delaying the progression of NASH, studies have shown that, for most individuals, it is generally very difficult to maintain over the long-term, even following bariatric surgery.
There are currently no drugs approved by regulatory authorities for the treatment of NASH. Even though certain drugs, such as insulin sensitizers and antihyperlipidemic agents, are prescribed for some NASH patients, they are not approved for the treatment of NASH and their efficacy has not been proven in adequate and well-controlled clinical studies.
Currently, it is impossible to predict which of the NAFLD patients will deteriorate to NASH as it is unclear what causes NASH to develop. Researchers are now focusing on several factors that may contribute to the development of NASH. Therefore, lifestyle changes are recommended for all patients with NAFLD.
There is an exceptionally wide range of estimates regarding the potential commercial market for NASH. This uncertainty stems from (i) the overall size of the patient population, (ii) the percentage of the addressable market that will be diagnosed and, subsequently, seek treatment, (iii) the ultimate cost of the therapies, (iv) the number of approved drugs for NASH and their profile, and (v) uncertainty regarding the regulatory approval process. Some of these factors cannot be known until NASH drugs begin to hit the market or biomarkers replacing the biopsy diagnosis are validated. Independent estimates generally estimate a commercial multi billion market in developed countries, though we do not endorse any estimates, which are based on a number of different underlying assumptions.
Aramchol for NASH
Overview
Our product candidate, Aramchol, is a first-in-class synthetic fatty acid-bile acid conjugate molecule, or FABAC, molecule that we are developing for oral treatment for NASH in patients who are overweight or obese and have prediabetes or type II diabetes mellitus.
Early in its development, Aramchol’s ability to modulate hepatic lipid metabolism was observed and validated in numerous pre-clinical trials with different animal species. Mice fed a high fat diet and treated with Aramchol did not develop fatty liver as compared to non-treated mice. In these early studies, we also observed that the mechanism of this effect was not a result of malabsorption of fat in the intestines because the FABAC-treated mice gained weight throughout the test periods to a similar degree to the control mice. This led us to conclude that FABAC therapy triggers a beneficial modulation of intra-hepatic lipid metabolism and reduces liver fat content.
| 59 |
In in-vitro and in vivo studies, Aramchol down regulates the SCD1 enzyme, an enzyme recognized as playing an important role in the metabolism of fatty acids. The SCD1 enzyme is essentially the gateway that regulates the use and storage of fat in the body by converting saturated fatty acids to monounsaturated fatty acids. Experimental animal studies showed that complete inhibition of the SCD1 enzyme protects against diet-induced obesity, hepatic steatosis, or fatty liver, and insulin resistance by instructing the body to use, rather than store, all fatty acids. However, various animal studies have indicated that such complete SCD1 enzyme inhibition has mechanism based serious side effects, such as atherosclerosis, and eye and skin disorders. As observed by us in our pre-clinical and clinical studies performed to date, and subsequently published in the European Journal of Gastroenterology and Hepatology and Archives of Medical Research in 2008 and 2010 respectively, one of Aramchol’s unique characteristics is that it down regulates the SCD1 enzyme but does not inhibit it completely – a partial effect. To date, side effects that have been observed in animals with knock out of SCD1 have not been observed in our toxicology and clinical studies.
To better understand the role of Aramchol in NASH, we analyzed the effect of Aramchol in MCD diet model. The aim of this study was to investigate Aramchol’s mechanism of action and its effect on fibrosis using the methionine- and choline-deficient (MCD) diet model of NASH. We collected liver and serum from mice fed a MCD diet containing 0.1% methionine (0.1MCD) for four weeks, which developed steatohepatitis and fibrosis, as well as mice receiving a control diet; the metabolomes and proteomes were determined. 0.1MCD fed mice were given Aramchol (5mg/kg/day for the last 2 weeks); liver samples were analyzed histologically. Aramchol administration was found to reduce features of steatohepatitis and fibrosis in 0.1MCD fed mice. Aramchol downregulated the SCD1 enzyme, a key enzyme involved in triglyceride biosynthesis whose loss enhances fatty acid β-oxidation. In addition, Aramchol increased the flux through the transsulfuration pathway, leading to a rise in glutathione (GSH) and GSH/GSSG ratio, the main cellular antioxidant that maintains intracellular redox status. Comparison of the serum metabolomic pattern between 0.1MCD-fed mice and patients with NAFLD showed a substantial overlap. These findings were published in Hepatology Communications, Vol. 1, No. 9, 2017.
As the effect of Aramchol on fibrosis was first reported we further analyzed the direct effect of Aramchol on collagen production and reported down regulation of collagen production from the hepatic stellate cells (HSCs) by Aramchol. With that we could conclude that Aramchol has potential direct effect on collagen production and therefore reduces fibrosis indirectly by down regulation of steatosis by reducing the sequence of events but also directly affecting collagen producing cells. These findings were published in Hepatology Communications, Vol. 1, No. 9, 2017.
These findings led us to further analyze the effect of Aramchol using the Thiocatemide (TAA) rat model. TAA is the most commonly used toxic agents to induce liver fibrosis. Repeated IP injections of TAA leads to sever fibrosis / cirrhosis. Among all models for fibrosis, the TAA model share multiple characteristics with human liver fibrosis and is considered to best predict efficacy in humans. Results demonstrated that treatment with Aramchol 5mg/kg, significantly prevented TAA induced fibrosis in a dose dependent manner. These findings were presented at EASL, Amsterdam in April 2017 (The anti Fibrotic effect of Aramchol on liver Fibrosis in TAA animal model).
Phase 1 Single and Multiple-Dose Study of Aramchol in Healthy Male Volunteers (NCT00776841)
Aramchol was evaluated in two Phase 1 clinical trials (under a single protocol) to study its safety, tolerability and PK profile in healthy volunteers, in both single and multiple dose administrations. The first Phase 1 clinical trial was an escalating single-dose trial conducted in 17 healthy subjects testing Aramchol doses ranging from 30 mg to 900 mg, performed in one center in Israel. The subsequent Phase 1 clinical trial was a repeated-dose trial conducted over four days in 25 healthy subjects testing repeated daily doses of Aramchol of 30 mg and 300 mg, performed in one center in Israel. The profiles for the groups were similar and the maximal plasma concentration of Aramchol increased with the higher doses. The PK profile demonstrated that Aramchol is suitable at each dose for once-daily administration and there were neither significant adverse events observed in either Phase 1 trial nor any notable changes in biochemical, hematologic, cardiovascular or other safety parameters.
| 60 |
Phase 2a Trial: Aramchol Treatment in NAFLD or NASH Patients (NCT01094158)
In January 2012, we completed a 60 patient multi-center, randomized, double-blind, placebo-controlled Phase 2a clinical trial of Aramchol in patients with NAFLD or NASH between the ages of 18 and 75 in 12 centers in Israel. The Phase 2a study results were published in July 2014 in the peer-reviewed Clinical Gastroenterology and Hepatology Journal. The trial was performed in patients with either NAFLD or NASH, which design was deemed acceptable by the FDA in 2007 at a pre-IND scientific advisory meeting. The trial’s primary efficacy endpoint was a reduction in liver fat content, and did not consider inflammation or fibrosis, which can be diagnosed only by liver biopsy. We believe that the short study duration of three months of treatment followed by a one-month follow-up period did not warrant repeated biopsies. The trial evaluated the effects on liver fat content of 100 mg and 300 mg once-daily doses of Aramchol compared to a placebo. At the end of the three month treatment period, statistically significant reductions in liver fat concentration as measured by MRS were observed in the 300 mg patient group. Specifically, a 12.57% mean liver fat content reduction was observed in the 300 mg group, as compared to a mean reduction of 2.89% in the 100 mg group and a mean increase of 6.39% in the placebo-treated patients. These results indicate that the effects of Aramchol are dose-dependent, as demonstrated in the graph below, which presents the results with respect to the 57 patients who successfully completed the entire treatment period (three patients were excluded from data analysis because of one protocol violation and two withdrawal consents).
Relative Change in MRS from Baseline after Three Months of Treatment
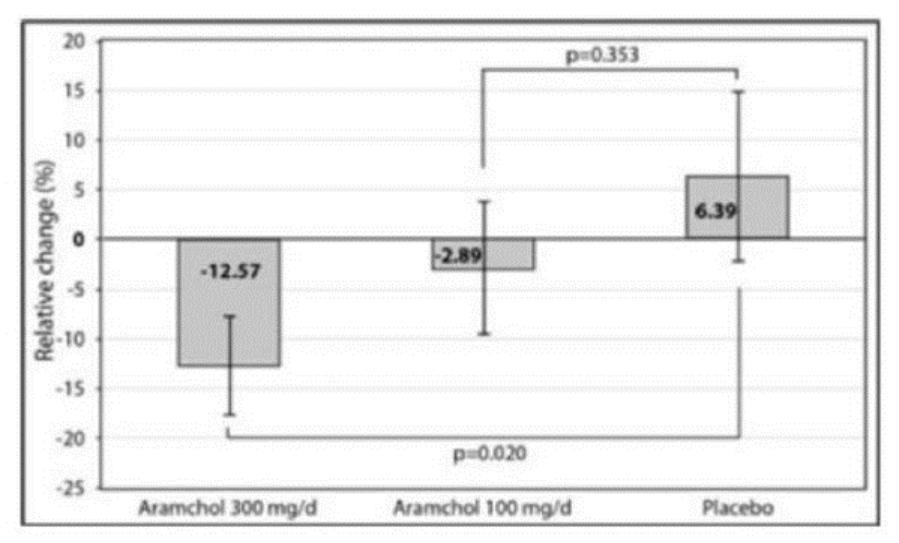
The table above shows that the primary endpoint of the study was attained. The study demonstrated a statistically significant, dose dependent reduction in fat content in the livers of patients treated with Aramchol, with a 19% difference between the 300 mg dose group and the placebo group, while the difference between the 100 mg dose group and the placebo group was not statistically significant. Notably, the minimal effective dose of Aramchol for fat reduction has been defined.
| 61 |
There were no statistically significant differences among the three treatment groups for any of the secondary end points. There was a non-statistically significant trend of mild weight reduction (P=.1) in the high dose Aramchol group. Serum adiponectin levels increased (0.2 ± 1.7 μg/mL) in the high-dose Aramchol group but decreased in the low-dose (-0.3 ± 1.5 μg/mL) and placebo groups (-0.7 ±_1.3 μgg/mL) (P= 0.88 for trend of dose-response relationship by linear regression). FMD increased non-statistically significantly by 1.28% ± 2.92% in the high-dose group, by 0.34% ±3.54% in the low-dose group, and by 0.46% ± 2.28% in the placebo group.
The frequency of adverse events was similar in all treatment groups, and none of them were considered to be related to the treatment. All adverse events in the active treatment arms were mild or moderate and none were serious. None of the patients withdrew as a result of adverse events. The following table shows the most frequent adverse events (occurring in ≥ 2 patients in any group) in the study.
| Placebo | Aramchol 100mg/d | Aramchol 300mg/d | ||||||||||||||||||||||||||||||||
| (N=20) | (N=20) | (N=20) | ||||||||||||||||||||||||||||||||
| MedDRA preferred term | No. Events | No. Subjects | % | No. Events | No. Subjects | % | No. Events | No. Subjects | % | |||||||||||||||||||||||||
| Abdominal pain | 2 | 2 | 10 | % | 2 | 1 | 5 | % | 1 | 1 | 5 | % | ||||||||||||||||||||||
| Abdominal pain upper | 1 | 1 | 5 | % | 2 | 2 | 10 | % | — | — | — | |||||||||||||||||||||||
| Constipation | 2 | 2 | 10 | % | — | — | — | — | — | — | ||||||||||||||||||||||||
| Asthenia | 2 | 2 | 10 | % | — | — | — | — | — | — | ||||||||||||||||||||||||
| Back pain | 3 | 3 | 15 | % | — | — | — | — | — | — | ||||||||||||||||||||||||
| Musculoskeletal pain | 2 | 2 | 10 | % | — | — | — | — | — | — | ||||||||||||||||||||||||
| Upper respiratory tract infection | — | — | — | — | — | — | 2 | 2 | 10 | % | ||||||||||||||||||||||||
The results of our Phase 2a clinical trial of Aramchol in the peer-reviewed Clinical Gastroenterology and Hepatology Journal were published in December 2014. The trial manuscript, entitled “The Fatty Acid-Bile Acid Conjugate Aramchol Reduced Liver Fat Content in Patients with Nonalcoholic Fatty Liver Disease,” provides the full report of the Phase 2a trial, which was completed in January 2012 and presented at the 47th Annual Meeting of the European Association for the Study of the Liver in 2012. Based on this Phase 2a proof-of-concept results, we established a development plan that we believe may confirm: (i) the good safety profile of Aramchol, (ii) the optimal dose of Aramchol, and (iii) efficacy on steatosis as well as fibrosis in patients with NASH.
Pharmacokinetics of Single and Multiple Escalating Doses of Aramchol and Food Effect in Healthy Volunteers (NCT02374437)
On April 28, 2014, we commenced PK and food effect studies of Aramchol. In written correspondence from December 2013 regarding a requested pre-IND meeting, the FDA recommended that we conduct such studies prior to commencing our Phase 2b ARREST Study.
We conducted the food effect and PK study at the Sourasky Medical Center in Tel Aviv, Israel involving 66 healthy volunteers to evaluate the PK of Aramchol following single and multiple escalating doses (200 mg, 400 mg and 600 mg), as well as to evaluate the effect of a high-fat, high-calorie meal on the PK of Aramchol following a single dose in healthy volunteers.
The results showed dose-related, but less than dose-proportional, increases in the mean Aramchol plasma concentrations, or Cmax, area under the curve, or AUC, (0-t), and AUC (inf) of 200 mg, 400 mg and 600 mg doses administered under fasting conditions or following a light meal, both at single and repeated dose administration. Cmax and AUC are metrics used to indicate the significance of a drug’s exposure. Steady-state was achieved by 144 hours (day seven). Administration of Aramchol after a high-fat, high-calorie meal afforded a 2.6 fold increase in exposure, as measured by Cmax, AUC(0-t), and AUC(inf) compared to the fasting group.
| 62 |
No serious adverse events or deaths occurred during the study. Adverse events were equally distributed between placebo and Aramchol doses, were mild (with only one moderate adverse event) and the majority defined unrelated to Aramchol. The PK study provides additional safety data to further support existing safety data from our pre-clinical studies and our Phase 1 and Phase 2a clinical trials of Aramchol.
Pharmacokinetics of Single and Multiple Escalating Doses of Aramchol Administered under Fed Conditions in Healthy Chinese Volunteers (NCT 02803996)
In 2016, we performed the Chinese PK Study involving Chinese patients who are domiciled in the United States. We enrolled 66 patients in this study, consisting of two parts. In part A, 32 subjects received a single escalating dose; Part B enrolled 34 subjects which received a multiple escalating dose. Dr. Evelyn Darius served as the Study Investigator. No safety signal was identified in this study and we deemed no changes were required in the enrollment of Chinese patients into the ARREST Study. Moreover, having this Chinese PK Study data may give us a head start in future licensing discussions with potential Chinese partners for the development of Aramchol in China.
Phase 2b ARREST Study for Aramchol (NCT 02279524)
In September 2014, the FDA granted Fast Track designation status to Aramchol for the treatment of NASH. Fast Track designation may accelerate the development process and may expedite the review of drugs that show promise in treating serious, life-threatening medical conditions for which no other drug either exists or is as effective.
On February 1, 2015, we began our ARREST Study. The ARREST Study was a Phase 2b, multicenter, global, randomized, double-blind, placebo controlled study to evaluate the efficacy and safety and of two doses of Aramchol for the treatment of NASH in patients who are overweight or obese and have pre diabetes or type II diabetes mellitus. In order to be eligible to participate in the ARREST Study, patients had to be affected by NASH, as diagnosed by a biopsy centrally read (steatosis ≥1 + inflammation ≥1 + ballooning ≥1, total activity NAS score of 4 or more), have a fibrosis stage of 1-3, be overweight or obese as measured by a Body Mass Index between 25 and 40 or waist circumference between 88cm to 200cm for women, and between 102cm to 200cm for men, and who are pre diabetic or type II diabetic. We targeted this specific population as it is at the greatest risk of developing NASH and its complications. We have generated data from animal models that lead us to believe that Aramchol targets all three main pathologies of the disease: steatosis, inflammation and fibrosis.
A total of 247 patients (approximately one third in the US, one third in Latin America and one third in Europe and Israel) with liver biopsy-proven NASH who were overweight or obese and had pre-diabetes or type II diabetes mellitus were randomized. Patients were randomized in a ratio of 2:2:1 (600mg, 400mg and placebo) taking once-daily oral Aramchol (in the Aramchol treatment arms) or a placebo (in the placebo arm). The treatment part of the trial was 12 months in duration and patients completing this phase were observed for a three month follow-up period. In February 2017, we completed randomization of the ARREST Study. Baseline histology of patients enrolled into the ARREST study demonstrated a population with advanced disease, with 60% having stage 2 and 3 fibrosis and 70% have NAS>5 at baseline.
The primary endpoint of the study was the change from baseline to end of study in liver triglycerides ratio as measured by magnetic resonance spectroscopy, or MRS (Aramchol 600mg vs. placebo). Secondary endpoints, demonstrated through biopsy, included fibrosis improvement by at least one stage or more without worsening of NASH (defined by an increase of inflammation and or ballooning) and NASH resolution (defined by ballooning score 0 and inflammation score 0-1 at termination) without worsening of fibrosis. Other secondary endpoints included improvement (2 points or more) in NASH activity index, as measured by NAS or SAF, without worsening fibrosis and change in baseline to week 52/termination in ALT (U/L).
| 63 |
On June 12, 2018, we announced top-line results of the ARREST Study and on November 13, 2018 an oral abstract presentation of one-year results of the ARREST Study was presented during a Late Breaking Abstract Oral Session at The Liver Meeting® 2018 during the American Association for the Study of Liver Diseases 2018 Annual Meeting.
Of the 247 patients, 48 patients were in the placebo arm, 101 patients in the Aramchol 400mg arm and 98 in the Aramchol 600mg treatment arm. The majority of subjects completed 52 weeks of treatment and 13 weeks of follow up (89.1%, 89.8%, 85.4% in the 400 mg, 600 mg and placebo arms, respectively). The leading cause of discontinuation was consent withdrawal and early termination due to adverse events; the incidence of early termination due to AEs was very low and similar across study arms.
Patients in the ARREST study were planned to undergo MRS, and a liver biopsy at baseline and week 52, which were centrally read, blinded to treatment allocation. The statistical analysis plan included pre-defined analysis sets: (i) a full analysis set for MRI (FAS -— MRI): all intent to treat, or ITT, patients with baseline and at least one second MRS. 214 patients were included in this analysis set (41 in placebo; 90 in Aramchol 400mg; and 83 in Aramchol 600mg); and (ii) a full analysis set for liver biopsy (FAS -— biopsy): all ITT patients with baseline and a second biopsy. 198 patients were included in this analysis set (40 in placebo; 80 in Aramchol 400mg; and 78 in Aramchol 600mg).
Results from the study showed a statistically significant reduction in liver fat by MRS with Aramchol 400mg vs. placebo (p=0.0450) and not with 600mg (p=0.0655) and thus did not reach the primary endpoint of the study. In a post-hoc analysis, a cutoff of 5% absolute reduction in liver fat was used as a surrogate for potentially clinically meaningful MRI reduction. In this responder’s analysis, a dose-response could be observed; the responder rate was 47.0%, 36.7% and 24.2%, in the Aramchol 600mg, 400mg and placebo arms, respectively. The proportion of the Aramchol 600mg arm compared to placebo was statistically-significant (p=0.0279).
Results for the two biopsy endpoints, which may currently constitute a primary endpoint for a Phase 3 trial to support an FDA marketing application, demonstrated the following: (i) significantly more patients treated with Aramchol 600mg vs. placebo achieved NASH resolution without worsening of fibrosis (16.7% vs. 5.0%; p=0.0514); and (ii) a higher proportion of patients showed at least one-point improvement in fibrosis score without worsening of NASH in Aramchol 600mg vs. placebo (29.5% vs. 17.5%; p=0.2110).
Statistically significant reductions in live enzymes alanine transaminase (ALT) and aspartate transaminase (AST) were demonstrated in both Aramchol arms vs. placebo (p≤0.0002) and (p<0.0001), respectively.
Secondary endpoints based on NAS and SAF activity score, ≥2 points improvement, showed a higher proportion of patients with improvement in the Aramchol arms (600mg>400mg>placebo; P>0.05).
Exploratory endpoints of glycemic parameters showed statistically significant reductions in HbA1c with both Aramchol arms vs. placebo (p<0.007) implying a potential effect on glycemic control.
At 52 weeks of treatment, Aramchol continued to show a favorable safety and tolerability profile. Serious adverse events were reported in 12.5%, 8.9% and 9.2% of patients in placebo, Aramchol 400mg and 600mg arms, respectively. No clustering of event type or atypical events for the studied population was reported in either Aramchol arms. Severe adverse events were reported in 10.4%, 6.9%, and 6.1% of patients in placebo, Aramchol 400mg, and 600mg arms, respectively. Early terminations due to adverse events occurred in 4.2%, 3.0% and 4.1% in placebo, Aramchol 400mg and 600mg arms, respectively.
| 64 |
The following table summarizes the most frequent adverse events.
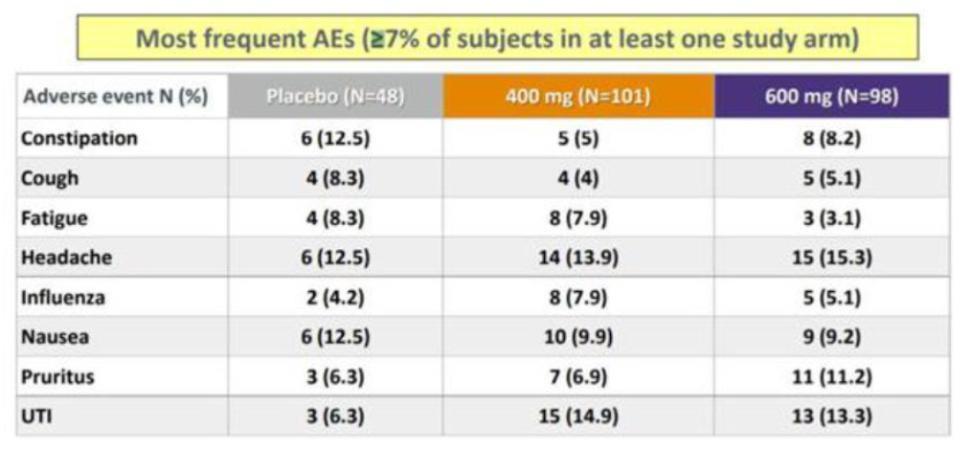
The following table summarizes the ARREST results:
| Aramchol | Aramchol | |||||||||||
| Placebo | 400mg | 600mg | ||||||||||
| MRS- –Absolute change from baseline in mean liver fat (1) | (0.09 | )% | (3.41 | )% | (3.18 | )% | ||||||
| P=0.0450 | P=0.0655 | |||||||||||
| MRS responders- Reduction of ≥5% in absolute change from baseline (1) | 24.4 | % | 36.7 | % | 47.0 | % | ||||||
| P=0.0878 | P=0.0279 | |||||||||||
| NASH resolution without worsening of fibrosis (2) | 5 | % | 7.5 | % | 16.7 | % | ||||||
| P=0.4955 | P=0.0514 | |||||||||||
| NASH resolution (2) | 7.5 | % | 12.5 | % | 19.2 | % | ||||||
| P=0.2237 | P=0.0462 | |||||||||||
| Fibrosis improvement (≥1 stage) without worsening of NASH (2) | 17.5 | % | 21.3 | % | 29.5 | % | ||||||
| P=0.8425 | P=0.2110 | |||||||||||
| Progression to Cirrhosis (Post-Hoc Analysis) worsening of NASH (2) | 7.5 | % | 7.5 | % | 1.3 | % | ||||||
| P=0.5693 | P=0.1008 | |||||||||||
| ALT (U/L) Change from baseline (3) | +11.82 | -12.0 | (17.3 | ) | ||||||||
| P=0.0002 | P<0.0001 | |||||||||||
| AST (U/L) Change from baseline (3) | +6.67 | (7.20 | ) | (10.83 | ) | |||||||
| p=0.0011 | p<.0001 | |||||||||||
| HbA1C Change from baseline (4) | +0.32 | (0.04 | ) | (0.13 | ) | |||||||
| p=0.0061 | p=0.0008 | |||||||||||
| (1) | Placebo N=41; 400mg N=90, 600mg N=83; Mixed Effect Model Repeat Measurement (MMRM) adjusted mean changes from baseline; p-values for comparison of active treatment arm vs. placebo. | |
| (2) | Placebo N=40, 400mg N=80, 600mg N=78; Baseline adjusted logistic regression; p-values for comparison of active treatment arm vs. placebo. | |
| (3) | Placebo N=47, 400mg N=100, 600mg N=98; MMRM adjusted mean changes from baseline; p-values for comparison of active treatment arm vs. placebo. | |
| (4) | Placebo N=47, 400mg N=98, 600mg N=96; MMRM adjusted mean changes from baseline; p-values for comparison of active treatment arm vs. placebo. |
| 65 |
Dose Splitting Pharmcokinetic Study (NCT03774173)
As a result of the dose response pattern observed in the ARREST Study, we recently conducted a Phase 1, open-label, crossover PK study to assess whether dose splitting of Aramchol 600mg to twice daily 300mg will significantly increase plasma levels. 16 healthy subjects took part in two study periods. Eight subjects received each regimen in the first period and the alternate regimen in the second period. A PK profile was obtained over the dosing interval at steady state on day ten of each period.
Results of the study showed that the administration of Aramchol 300 mg twice daily resulted in 24-hour plasma concentrations significantly greater than those observed with the administration of Aramchol 600 mg once daily. (P<0.0001). The average plasma levels (exposure) were 53% higher and exposure was greater in all 16 subjects with the twice daily dosing. The treatment in both dosing regimens were similar in terms of safety and were well tolerated.
First in Human Aramchol Meglumine Pharmcokinetic Study
In December 2020, we announced new data from a Phase 1, first in human study that compared Aramchol meglumine to Aramchol acid. Armachol acid and Aramchol megluine was administered twice daily to 12 subjects. Below is a summary of the results:
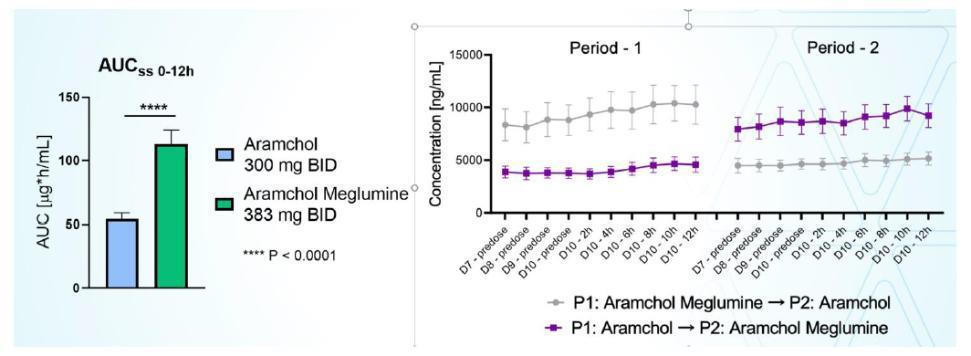
These initial results demonstrated that the new salt form of Aramchol meglumine has a plasma PK profile that is very similar to Aramchol acid. It also showed that the administration of both forms resulted in the same form of Aramchol in the blood, regardless of which drug product is administered and that less Aramchol meglumine is needed for the same exposure of Aramchol acid in the blood.
| 66 |
Phase 3 ARMOR Study for Aramchol
In September 2019, we initiated the ARMOR Study, a Phase 3 pivotal study of Aramchol for the treatment of NASH, following a successful End-of-Phase 2 meeting with the FDA in April 2019 in which we reached general agreement on key aspects of the Phase 3 development and registration plan for Aramchol. The ARMOR Study was originally comprised of two parts, a randomized, double-blind, placebo-controlled histology -based registrational part where 1200 subjects will be treated with Aramchol or matching placebo for 52 weeks and a clinically based part where subjects will continue with the same treatment for approximately five years, taking into consideration draft guidance issued by the FDA in December 2018 entitled “Noncirrhotic Nonalcoholic Steatohepatitis with Liver Fibrosis: Developing Drugs for Treatment”, or the “December Guidance”. The histology-based data is intended to serve as the basis for the submission of a marketing authorization application under regulatory provisions of Sub-part H accelerated/conditional approval.
In light of the rapid development of the Aramchol meglumine program and due to the delays resulting from the COVID-19 pandemic, in December 2020, we announced the addition of an open label part to the ARMOR Study and temporarily suspended randomization of new patients into the double-blind, placebo-controlled histology-based registrational phase of ARMOR as currently enrolled patients are transitioned to the open label part.
The following is a summary of the clinical trial design of ARMOR:
The Phase 3 study is a two-part study, an open-label part and a randomized, double-controlled, placebo part, designed to evaluate the safety and efficacy of Aramchol and to be conducted in approximately 200 sites in the U.S., Europe and Latin America.
Part One: Open Label Study
The first part, an open-label study, was originally designed to evaluate treatment response kinetics, pharmacokinetics and safety of twice daily administration of Aramchol 300mg in approximately 150 subjects with NASH and liver fibrosis stage 1-3 (F1 capped at 30 subjects), subjects with NASH who may or may not be overweight, and subjects with NASH who may or may not have type 2 diabetes or be pre-diabetic. Patients were randomized (1:1:1) into three groups with post-baseline liver biopsy being performed at 24 weeks, 48 weeks, or 72 weeks, respectively. A second post-baseline liver biopsy was conducted after one year for subjects whose post-baseline liver biopsy at week 24, 48 or 72 does not show at least one stage improvement in fibrosis. The open label part was being conducted at approximately 50 selected sites in the U.S., and around the world which have been less affected by the COVID-19 pandemic.
The second part, a randomized, double-blind, placebo-controlled study, is designed to evaluate the safety and efficacy of twice daily administration of Aramchol 300 mg to support regulatory approval, with both a histology-based phase and a clinically-based phase. As currently designed, a total of 2000 subjects with NASH and liver fibrosis stage 2 and 3 who are overweight and are either pre-diabetic or have type 2 diabetes are expected to be randomized 2:1 to receive Aramchol 300mg BID or matching placebo. In the histology-based phase, we intend to treat 1000 subjects with Aramchol or matching placebo for 72 weeks until the second biopsy. The histology-based data is intended to serve as the basis for the submission of a Sub-part H marketing authorization application under regulatory provisions of accelerated/conditional approval. The primary histology-based endpoint is NASH resolution without worsening of fibrosis or fibrosis improvement without NASH worsening. In the clinically-based phase, all subjects will continue with the same treatment assignment for up to seven years until study completion to confirm clinical efficacy. We may announce end-of-study at the time when a total of 380 subjects have experienced at least one pre-specified clinical event or at five years from last subject randomization, whichever comes first. The primary clinically-based endpoint is expected to be based on clinical events including all-cause mortality, histological progression to cirrhosis, MELD score >15, and hepatic decompensation events (e.g., hepatic encephalopathy, variceal bleeding, ascites).
| 67 |
In August 2021, we announced that the FDA agreed with our plan to use Aramchol meglumine (in lieu of Aramchol free acid) in our ARMOR Study without the need to conduct additional nonclinical and clinical studies other than planned limited pharmacology studies relating to Aramchol meglumine. In addition, the Medicines and Healthcare products Regulatory Agency, or MHRA, the pharmaceuticals regulator in the UK, also agreed with our plan to proceed with our proposed clinical studies with Aramchol meglumine in lieu of Aramchol free acid without the need to repeat nonclinical and clinical studies other than planned limited pharmacology studies relating to Aramchol meglumine.
In November 2021, we announced positive interim data on the open label part of the ARMOR Study showing clinically significant effect on fibrosis improvement based on histology in the first 16 patients. Subsequently in November 2021, we announced that new analyses of biomarkers corroborate this effect showing statistically significant reductions in biomarkers associated with liver fibrosis including ALT, AST, Fib-4 and ProC-3. Reductions of a similar magnitude are seen in a cohort of the first 20 patients for which paired biopsy have been analyzed (Late breaker AASLD Cohort N=20) and a cohort of 50 patients for which biomarker data was analyzed (ARCON Cohort N=50) based on all available data (N=139). Aramchol continued to show excellent safety and tolerability profile. Data support that higher dose of Aramchol could provide statistically and clinically meaningful effect on fibrosis in the upcoming double-blind placebo controlled part for submission of the ARMOR study to support an NDA under Sub-part H. Moreover, we believe the results supported discussions with FDA to potentially allow a smaller and shorter double-blind placebo control histology-based part to support regulatory submission of an NDA.
A summary of the results is presented below:
| Late Breaker AASLD Cohort 1 (N=20) | ARCON Cohort 2 (N~50) | |||||||||||||||||||
| visit | Change from baseline 3 | p value | Change from baseline 3 | p value | ||||||||||||||||
| week 24 | -20.88 | <.0001 | -16.44 | <.0001 | ||||||||||||||||
| ALT | week 48 | -20.05 | <.0001 | -17.43 | <.0001 | |||||||||||||||
| week 24 | -15.08 | <.0001 | -13.43 | <.0001 | ||||||||||||||||
| AST | week 48 | -14.74 | <.0001 | -13.28 | <.0001 | |||||||||||||||
| week 24 | -0.22 | 0.0006 | -0.27 | <.0001 | ||||||||||||||||
| FIB-4 | week 48 | -0.23 | 0.0012 | -0.22 | 0.0006 | |||||||||||||||
| week 24 | -8.83 | 0.0010 | -9.68 | <.0001 | ||||||||||||||||
| PRO-C3 | week 48 | -13.79 | 0.0005 | -13.00 | <.0001 | |||||||||||||||
| week 24 | -15.89 | 0.0106 | -13.84 | 0.0077 | ||||||||||||||||
| PRO-C3 % | week 48 | -19.76 | 0.0294 | -17.37 | 0.0192 | |||||||||||||||
| >1 point in fibrosis improvement | 60 | % | ||||||||||||||||||
| 1. | Number of subjects in the AASLD cohort at week 24 and week 48 respectively are; ALT 19 and 9, AST 19 and 9, FIB-4 19 and 9, PRO-C3 19 and 10 |
| 2. | Number of subjects in the ACRON cohort with data at week 24 and week 48 respectively are; ALT 61 and 18, AST 61 and 18, FIB-4 56 and 18, PRO-C3 43 and 15 |
| 3. | MMRM baseline adjusted analysis |
| 4. | ProC-3 were analyzed by Nordic Bioscience using an improved methodology with higher sensitivity and specificity. Mean Baseline 47.2 ug/L. |
| 68 |
In April 2022, we announced further positive interim data on the open label part of the ARMOR Study showing robust fibrosis improvement across multimodality histological assessment. Biopsies were read by three independent pathologists individually, followed by a consensus reading. The same central committee were also asked to perform a ranked assessment (improvement/worsening/stable) of paired (pre and post baseline) biopsies scrambled and blinded to sequence. an automated and continuous score of Fibrosis Composite Severity (FCS) was established for the same slides using FibroNest™, a quantitative Digital Pathology image analysis and artificial intelligence (AI) method. Results of post baseline biopsies performed either at 24 weeks or at 48 weeks from 46 subjects with NASH and F1-3 that received Aramchol support the anti-fibrotic effect of Aramchol and reinforce the favorable safety profile of Aramchol.
A summary of the results is presented below
| Biopsy Methodologies | 24 Weeks | ≥ 48 Weeks | ||||||||||||||
| N | % | N | % | |||||||||||||
| All | 26 | 100.0 | 20 | 100.0 | ||||||||||||
| Fibrosis Improvement (1 stage or more) based on NASH CRN | 7 | 26.9 | 8 | 40.0 | ||||||||||||
| Fibrosis Improvement (Paired reading ranked assessment) based on comparing individual patients slides | 11 | 42.3 | 13 | 65.0 | ||||||||||||
| Subject Fibrosis Response (AI reading) using Fibronest’s Phenotypic Fibrosis Composite Score (A responder is defined by an absolute reduction of > 0.3 units) | 15 | 57.7 | 20 | 100.0 | ||||||||||||
Treatment with Aramchol 300mg BID resulted in a high rate of subjects with fibrosis improvement across the three pathology reading methods. Both paired and AI evaluations identified more subjects with fibrosis improvement, indicating greater sensitivity to detect change. For all methods, a treatment effect was larger at 48 compared to 24 weeks. AI analysis showed mean FCS reduction was -0.62 (p=0.017) at Wk24 and -1.74 (p<0.0001) at Wk>48.
Part Two: Histology-Based and Clinically-Based Study
The second part of the ARMOR Study is a randomized, double-blind, placebo-controlled study to evaluate the safety and efficacy of Aramchol 300 mg BID to support regulatory approval, with both a histology-based phase and a clinically-based phase. As currently designed, a total of 2000 subjects with NASH and liver fibrosis stage 2 and 3 who are overweight and are either pre-diabetic or have type 2 diabetes would be randomized 2:1 to receive Aramchol 300mg BID or matching placebo. In the histology-based phase, we intend to treat 1000 subjects with Aramchol or matching placebo for 72 weeks until the second biopsy. The histology-based data is intended to serve as the basis for the submission of a Sub-part H marketing authorization application under regulatory provisions of accelerated/conditional approval. The primary histology-based endpoint is NASH resolution without worsening of fibrosis or fibrosis improvement without NASH worsening. In the clinically-based phase, all subjects will continue with the same treatment assignment for up to seven years until study completion to confirm clinical efficacy. We may announce end-of-study at the time when a total of 380 subjects have experienced at least one pre-specified clinical event or at five years from last subject randomization, whichever comes first. The primary clinically-based endpoint is expected to be based on clinical events including all-cause mortality, histological progression to cirrhosis, MELD score >15, and hepatic decompensation events (e.g., hepatic encephalopathy, variceal bleeding, ascites). If the clinical trial results in the histology-based phase are positive, we plan to submit an NDA for Sub-part H accelerated/conditional approval to the FDA. As discussed elsewhere, we suspended the initiation of the double-blind placebo-controlled histology-based registrational part of ARMOR and are seeking to initiate the double-blind placebo-controlled histology-based registrational part of ARMOR in the second half of 2023, subject to, among other things, the results of our open label part, sufficient funding and clarification of the regulatory approval process for NASH drugs.
| 69 |
The following is a depiction of part two of the ARMOR Study:
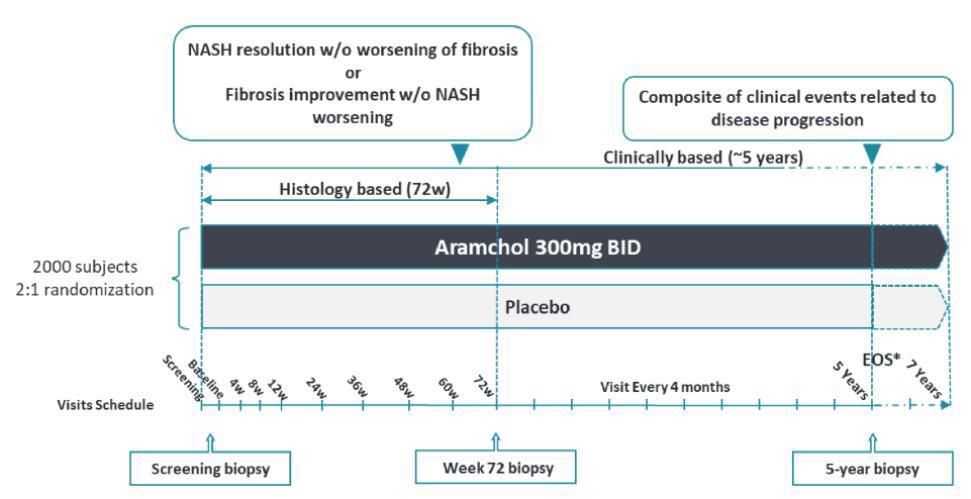
Amilo-5MER
In August 2020, we announced significant progress in the development of Amilo-5MER, a 5 amino acid synthetic peptide MTADV (Methionine, Threonine, Alanine, Aspartic acid, Valine). The 5 amino acids sequence of Amilo-5MER is homologue to a specific MTADV sequence in the human CD44 variant found in synovial fluid cells from joints of rheumatoid arthritis, or RA patients.
Amilo-5MER is being developed through a research collaboration between us and the Hebrew University of Jerusalem. The molecule originated in the laboratory of Prof. David Naor, from the Lautenberg Center for Immunology and Cancer Research, Faculty of Medicine, The Hebrew University. Prof. Naor and his team were the first to publish this specific sequence in the prestigious scientific communication Journal of Clinical Investigation 1.
Amilo-5MER binds to three pro-inflammatory amyloid proteins, Serum Amyloid A, or SAA, Transthyretin and Apolipoprotein B with high affinity. The first two are known to be active only in their aggregated forms. By binding to SAA, Amilo-5MER interferes with SAA aggregation and therefor inhibits the destructive autocrine, self-amplifying cytokine loop that causes additional inflammatory reaction.
SAA constitutes acute phase reactants, whose concentration in serum rise rapidly in response to acute stimuli such as infection and trauma. An elevated concentration of SAA was identified in sera of patients with multiple autoimmune diseases and more recently, an outstanding increase of SAA was also detected in COVID-19 infected patients2-3. SAA in its aggregated form, is a potent and rapid inducer of cytokine secretion (particularly Interleukin 6 (IL-6). IL-6 plays an important role in chronic inflammation and is implicated in the pathogenesis of many autoimmune diseases, such as Multiple Sclerosis, or MS, RA, Inflammatory Bowel Disease, or IBD and acute COVID 19. Interference with SAA polymerization and aggregation is a valid target to prevent chronic inflammatory conditions.
| 70 |
Amilo-5MER has been shown to significantly reduce chronic inflammation in animal models of RA, IBD and MS (research work supported by a grant to Prof. Naor from the National Multiple Sclerosis Society (NMSS) of the USA). Amilo-5MER provides a unique mechanism of action to interfere with this vicious cycle, enabling a specific treatment for chronic inflammatory diseases. Data generated from multiple in-vitro, in-vivo and human ex-vivo models have shown that Amilo-5MER significantly improves clinical symptoms. Histological improvements and reduction of pro-inflammatory cytokine secretion were also observed.
Amilo-5MER is considered a New Chemical Entity. As such, it is eligible for NCE patent protection until July 2034. Patents have been granted and maintained in the US (US 1061181937), Europe (EP 3169343) and Australia (AU 2015291151) and have been allowed in Japan (JP 6671363).
In March 2021, we dosed the first subject in our first in human Phase 1 trial of Amilo-5MER for the treatment of chronic inflammatory diseases. The trial was a three-part, single center, double-blind, randomized, placebo-controlled first in human study of single ascending doses (Part 1) and multiple doses (Part 2) of Amilo-5MER in young healthy adult male subjects and a single dose cohort in healthy elderly male and female subjects (Part 3). Overall, 55 healthy male and female subjects were enrolled in the study. Cohorts of 8 subjects were randomized to receive Amilo-5MER or placebo by subcutaneous injection in a ratio of 6:2. In Part 1, cohorts of young male adults received single ascending doses of 10, 30, 90, 180 and 360 mg; in Part 2, a single cohort received doses of 180 mg BID for 5 consecutive days and in Part 3 a single cohort of healthy elderly male and female subjects received a single dose of 180 mg. The primary objectives of the trial were to evaluate the safety, tolerability, and pharmacokinetics of Amilo-5MER. In January 2022, we announced results in which all doses of Amilo-5MER were well tolerated with no clinically significant adverse events and none considered related to the investigational product. All subjects completed the study as per protocol. Overall exposure to Amilo-5MER increased with an increase in dose, with statistically significant dose proportionality over the 10 mg to 360 mg dose range.
Additional Pre-clinical and Clinical Studies Required for Regulatory Submissions
Toxicology Studies
Since the completion of the Phase 2a study, pre-clinical toxicology studies have been conducted to support our ongoing clinical programs and regulatory submissions. These studies were performed in compliance with the EMA’s ICH M3 (R2) guidelines. The toxicity program for Aramchol included repeat dose studies of up to six months in rats and up to nine months in dogs by oral administration, the intended route of administration in the clinical trials and beyond. The dose level of 1000 mg/kg/day in rats and 1500 mg/kg/day in dogs, which is the maximal feasible dose in both species showed no side effect and therefore the highest dose of the study was selected as the no-observed-adverse-effect-level, or NOAEL. There were no observations noted in the rat study. The findings in the dog study were limited to changes in plasma lipids, including decreases in total blood cholesterol levels, LDL, HDL and phospholipids, and a slight increase in the size of the adrenal glands, which were considered to be an extension of the primary pharmacology of Aramchol and non-toxic effects, and skin scales from week 13 onwards in all Aramchol-treated groups, with a dose-related incidence. After six months this was not accompanied by any microscopic alteration of the skin and therefore considered not toxicologically relevant. Results from the study show that after nine months the presence of scales in all Aramchol-treated groups was accompanied by minor test item-related microscopic findings in the skin: Hyperkeratosis of the epidermis, correlating to the scales, and keratin plugs in the hair follicles (in males at 750/500 and 1500 mg/kg). After a 12-week treatment-free recovery period, fewer scales were noted and microscopically there was partial recovery. As these findings were minor and no clinical symptoms like scratching were noted, these findings were considered not adverse.
Aramchol was non-mutagenic in vitro in the Ames test and chromosomal aberrations test, each of which is a test to determine whether the subject chemical can cause mutations in the DNA of an organism. In addition, in bone marrow micronucleus test in male rats at a 2000 mg/kg oral dose (the maximum recommended dose in accordance with ICH S2 (R1)), Aramchol was not clastogenic, meaning it did not give rise to or induce disruption or breakages of chromosomes, nor was it aneugenic, meaning it did not cause the number of chromosomes in the nucleus of a cell to not be an exact multiple of the monoploid number of a particular species.
| 71 |
Embryo-fetal development toxicity was assessed in rats and rabbits. No maternal or fetal development toxicity was observed in either species. The NOAEL for maternal and development toxicity was at least 1000 mg/kg in rats and 750 mg/kg in rabbits (the maximum feasible dose in both species).
No maximum tolerated doses were reached in the studies. Over 50-fold safety margin exposure was achieved in dogs but not in rats. However, for rats, at least three of the four ICH M3(R2) safety margin criteria were met, and for dogs all four criteria were met. Blood tests revealed a decrease in total blood cholesterol levels, including LDL, HDL and phospholipids, and there was a slight increase in the size of the adrenal glands of the dogs, which WIL Research assessed as a physiologic compensatory response to the decrease in blood cholesterol levels. WIL Research did not consider the decrease in blood cholesterol levels or the physiologic response of the adrenal glands as a toxic effect, but rather as a pharmacodynamic effect, which is a biochemical and physiological effect of the drug on the body. Based on the above, it was concluded that the overall safety data for Aramchol is sufficient to support the proposed Phase 2b clinical trial.
To support any potential future NDA, we have commenced or are planning to commence several safety studies. Additionally, we plan in the future to conduct a study of Aramchol in the pediatric population.
In addition, we are conducting carcinogenicity studies to identify whether Aramchol has any tumorigenic potential upon long-term administration in support of any future NDAs or MAAs. Under FDA guidance, we are required to perform two studies, one in rats and the other in mice. The carcinogenicity study in rats is a two-year study which was initiated in February 2020.
Aramchol for the Treatment of Other Indications
On February 14, 2018, we announced topline results from the investigator initiated ARRIVE Study for HIV associated lipodystrophy and NAFLD patients. HIV patients have advanced liver disease which is a major cause for morbidity and mortality. ARRIVE, a Phase 2a, investigator initiated clinical trial conducted at the University of California
San Diego by Professor Rohit Loomba was a randomized, double-blinded, placebo-controlled, 12 weeks, proof-of-concept study that evaluated the safety and efficacy of Aramchol at 600mg/day versus placebo in 50 patients with HIV-associated lipodystrophy and NAFLD. The primary end point of successful therapy was improvement in hepatic steatosis at 12 weeks, as measured by MRI-PDFF. Secondary endpoints were improvement in total body fat, metabolic profile, and liver biochemistry. Liver biopsies were not included as part of the evaluation in this pilot trial. The trial showed no difference between HIV patients receiving Aramchol for 12 weeks when compared with HIV patients in the placebo arm. Aramchol showed a favorable safety and tolerability profile. Although the pathology (fatty liver) is similar to “garden variety” NASH, the pathogenesis involved in the HIV lipodystrophy and NAFLD is different and multi factorial including the effect of the virus itself and the anti-HIV medications.
On November 13, 2014, we announced the first administration of Aramchol in a proof-of-concept Phase 2a clinical trial for the treatment of newly formed cholesterol gallstones following bariatric surgery. The primary end-point was to prove that Aramchol dissolves newly formed gallbladder gallstones following bariatric surgery. Patients were to be assigned to one of three treatment arms; 400mg tablets, 600mg tablets and placebo. Only 9 patients were enrolled, and 7 patients completed the study. Due to poor patient recruitment and change in Company focus, we decided to terminate the study on October 1, 2015. We currently believe that it is unlikely that we will revive another study in cholesterol gallstones.
| 72 |
Our Competitive Strengths
We believe our key competitive strengths include the following:
| ● | A drug that targets the main NASH pathologies; steatosis, inflammation and fibrosis. We have generated data from animal models that lead us to believe that Aramchol targets all three main pathologies of NASH: steatosis, inflammation and fibrosis. The effect of Aramchol on fibrosis has shown to be indirect via reduction of steatosis and ballooning, and direct via reduction of collagen production from human hepatic stellate cells, the principle fibrogenic cell in hepatic fibrosis, and therefore has a potential to show significant results in NASH resolution without fibrosis worsening and/or fibrosis improvement without worsening of NASH. | |
| ● | 600mg dose of Aramchol in ARREST Study demonstrated a significant effect on an endpoint that may currently constitute a primary endpoint for a Phase 3 trial to support an FDA marketing application. In our Phase 2b ARREST Study, significantly more patients treated with Aramchol 600mg vs. placebo achieved NASH resolution without worsening of fibrosis (16.7% vs. 5.0%; p=0.0514). Under current FDA guidance, resolution of NASH and no worsening of liver fibrosis on NASH may currently constitute one of two endpoints that support an FDA marketing application. We believe that if we observe a similar effect on patients in our ARMOR Study, then we believe Aramchol is well positioned to be approved by the FDA. Moreover, in a dose splitting study of 300mg administered twice daily, we observed significantly higher exposure which suggests a potential for even higher efficacy with higher exposure of Aramchol. | |
| ● | An orally delivered drug with a good safety profile. In its current formulation, Aramchol is administered orally as a tablet. Simple and convenient oral delivery is expected to lead to increased patient compliance. Together with Aramchol’s good safety profile, we believe that Aramchol is well positioned against the competition in the treatment of NASH, where some treatments under development may require intravenous delivery or may cause adverse events, such as itching or an increase in LDL, which can be highly inconvenient for patients with chronic diseases, such as NASH, and may result in low patient compliance. If approved, Aramchol may enable physicians to treat NASH patients with moderate to severe fibrosis in all stages of NASH for long periods of time. | |
| ● | Experienced team with extensive knowledge and expertise in drug development. The Galmed team is highly skilled, experienced, and professional, which enables product development in an efficient, cost effective manner to enable timely regulatory approval. We believe our management team, scientific advisors and personnel have extensive knowledge and experience in the treatment of liver diseases, developing FABACs, such as Aramchol, for the treatment of liver diseases and working with lipid molecules, which due to their special physiochemical characteristics, are difficult to synthesize, develop and work with. We believe that such knowledge and expertise makes us competitive in the fields of metabolic and liver diseases. |
Our Strategy
Our strategy is to build a specialized biopharmaceutical company that develops, in a cost-effective manner, novel molecules from clinical stage to market readiness. We seek to create global partnerships with academic institutions and biotechnology or pharmaceutical companies to effectively collaborate in developing a portfolio and ultimately out-license Aramchol. Through this approach, we have successfully advanced Aramchol into various stages of clinical development. Key elements of our strategy include:
| ● | Continue advancing Aramchol through development as a first-in-class treatment for NASH and fibrosis. Following the completion of our Phase 2b ARREST Study, we are advancing Aramchol into a Phase 3 ARMOR Study with the goal of offering a first-in-class treatment for NASH. |
| 73 |
| ● | Explore strategic partnerships for Aramchol in different geographies. We seek to strategically partner with pharmaceutical and healthcare companies that possess experience, resources and infrastructure to execute clinical trial(s), regulatory approval and/or market launch. As part of this strategy, in July 28, 2016, we signed a license agreement with Samil for the commercialization of Aramchol in Korea. See “Item 4. Information on the Company—Business Overview—Strategic Collaborations, Research Arrangements and Other Material Agreements—Samil Pharm. Co., Ltd.” for more information regarding the Samil Agreement. In addition, we are actively exploring strategic partnership opportunities in other regions. | |
| ● | Investigate possible therapeutic combinations of Aramchol with drugs manufactured by others. We are seeking to co-develop Aramchol as a best in class drug with drugs manufactured by others in order to increase the commercial opportunities of Aramchol. | |
| ● | In-license, develop or acquire additional drug candidates. To diversify and expand our product pipeline, we are currently evaluating the acquisition or in-licensing of additional product candidates and technologies. |
Strategic Collaborations, Research Arrangements and other Agreements
Samil Pharma. Co., Ltd.
On July 28, 2016, we entered into a license agreement, referred to herein as the Samil Agreement, with Samil for the commercialization of Aramchol (with the option to manufacture) in the Republic of Korea, or the Territory.
Under the terms of the Samil Agreement, the Company has granted Samil an exclusive licence, or the Samil License, for fatty liver indications including NASH, or the Field of Use, in the Republic of Korea, or the Territory to such information concerning Aramchol as may be required to support Samil’s applications for regulatory approvals, or the Licensed Information, and the patents for the import, marketing, use, sale, offer for sale, commercialisation and distribution (and, if the option is exercised, manufacture) of Aramchol in tablet form, or any other physical form as may be produced or manufactured by or on behalf of Galmed or by a third party for Galmed and, if the option set out below is exercised, any products within the Field of Use, the development, manufacture or sale of which is based, in whole or in part, on, or involves the use of, the Licensed Information or covered under any patent, or the Product.
The Samil License shall remain in force with respect to each Product (if the Samil Agreement is not early terminated) until the later of: (i) the date of expiry in the Territory of the last of any patent covering such Product or any formulation, dosing or administration form thereof; and (ii) the date of expiry of a period of 20 years commencing on the date of first commercial sale by Samil or a sublicensee of such Product in the Territory.
Upon the signing of the Samil Agreement, Samil paid the Company a gross upfront fee of approximately $2.1 million and in September 2018, we received a milestone payment of $1.5 million. Samil has also agreed to pay additional clinical and regulatory-based milestone payments, which may aggregate to an additional $4.5 million, as well as tiered, double-digit royalties payable on sales (lower if sales of a generic equivalent commence in the Territory).
Pursuant to the terms of the Samil Agreement, following the first achievement of US$25 million of net sales in any calendar year following the first commercial sale of the Product in the Territory, Samil shall have the option to request that the Licensed Information include methods for the formulation of Aramchol from its API, to allow for the manufacture of Aramchol by Samil; provided, however, that we shall have the option, to widen the definition of the Licensed Information as aforesaid at any time.
| 74 |
We shall be entitled, at our option: (i) to modify the Samil License with respect to any Product so that it is non-exclusive only; or (ii) to terminate the Samil License hereunder, with respect to any Product if: (a) a first purchasing order from Samil for at least one Product shall not have been placed by 6 months following the grant of the Korean Ministry of Food and Drug Safety new drug approval; or (b) commercial sale of such Product having commenced and either (i) there shall be a period of 1 year during which no sales of any Product shall take place, or (ii) within 1 year of such commencement, aggregate sales of Products shall not have reached a reasonable level, as determined by the joint development committee, in each case, except as a result of force majeure or other factors beyond the control of Samil. Further, we shall be entitled to terminate the Samil Agreement if Samil challenges the validity of any of the patents. If any such challenge is unsuccessful, Samil shall (in addition to our right to terminate) pay us liquidated damages in the amounts of US $8,000,000. Either party may terminate the Samil Agreement (i) upon the other party’s material breach if such party fails to cure such breach within 30 days, or, in the case of failure by Samil to pay any amount due from Samil to us pursuant to or in connection with the Samil Agreement 14 days after receiving written notice thereof, or (ii) upon customary events such as the granting of a winding-up order if such order or act is not cancelled within 60 days.
In the event that we do not achieve the primary endpoint as defined in the study protocol, or Successful Completion, of the ARREST Study, we shall as soon as practicable notify Samil of the non-achievement of such Successful Completion, and within 60 days thereof, notify Samil in writing either: (i) that we have decided not to develop the Licensed Information further for the Field of Use, or the Cessation Notice, or (ii) that we intend to continue with such development notwithstanding the non-achievement of such Successful Completion, or the Licensor Continuation Notice. Also, in the event that we do not achieve the Successful Completion of the potential Phase 3 Study, we shall, as soon as practicable, notify Samil accordingly, or the Notice of Non-Success. Samil shall thereafter have the option, by notice in writing served to us within 45 days of Samil’s receipt of either a Cessation Notice, a Licensor Continuation Notice or a Notice of Non-Success, as applicable, to indicate its intention either: (i) to terminate the Samil License, or (ii) to continue research and development of the Licensed Information in the Field of Use in the Territory, or the Licensee Continuation Notice. In the event Samil shall serve a Licensee Continuation Notice following the service of a Cessation Notice or a Notice of Non-Success, any such continuation by Samil shall be subject to the entry by Samil into a written agreement with us as to the terms and conditions which would govern such continued research and development, which would be carried out according to Samil’s own development plan and at its sole expense. In the event Samil serves a Licensee Continuation Notice following the service of a Licensor Continuation Notice, or Agreed Continuation, the Samil Agreement shall continue in accordance with its terms. In August 2018, Samil sent a Licensee Continuation Notice to us.
Additionally, following the Successful Completion of the ARREST Study or Agreed Continuation following non-achievement of Successful Completion of the ARREST Study, Samil shall, for a period of 90 days following the date of written notification to it by us of such Successful Completion or following the date of Agreed Continuation following non-achievement of Successful Completion, have the option to require that the Territory be extended to include Vietnam, or the Extension Option. In the event that Samil exercises its Extension Option, the parties shall conduct negotiations in good faith for up to 30 days thereafter in order to agree on milestone payments which would replace those set out in the Samil Agreement. In the event that agreement is not reached in such regard within such period, the Extension Option shall terminate. Discussions for the extension of the Samil License to Vietnam are ongoing.
Amilo 5-MER
We have entered into a research and option agreement with Yissum, the tech transfer company of the Hebrew University with respect to our Amilo-5MER, a 5 amino acid synthetic peptide MTADV (Methionine, Threonine, Alanine, Aspartic acid, Valine). Under this agreement, we are able to research and initially develop Amilo-5MER, are required to fund the initial research and have been granted an exclusive option to negotiate and enter into a definitive license agreement with Yissum for Amilo-5MER upon certain pre-agreed upon terms and such other terms to be agreed upon.
| 75 |
As a result of the success in the Phase 1 study, we exercised our option and on June 28, 2021, we entered into a license agreement with Yissum pursuant to which Yissum granted us a worldwide, exclusive and irrevocable license to develop and commercialize Amilo-5MER. In November, 2021, the grant of the license took effect when it was approved by the Israel Innovation Authority. Under the license agreement, we are responsible for carrying out the development and commercialization of Amilo-5MER and the prosecution and maintenance of the licensed patents under the license agreement. In consideration for the grant of the license, we have agreed to pay to Yissum an upfront license fee of $100,000, payments of up to $950,000 upon meeting certain regulatory milestones, single digit royalties on any future net sales and a share of any sublicense fees. Unless earlier terminated, the license will continue in effect on a product-by-product and country-by-country basis until the later of (i) the expiration of the last to expire patent covering the licensed technology in such country, (ii) the expiration of any exclusivity on Amilo-5MER granted by a regulatory body in such country, and (iii) 15 years from the first commercial sale in such country. The license agreement may be terminated early for material breach or bankruptcy. In addition, we may terminate the license agreement without cause upon 90 days prior written notice to Yissum and Yissum may terminate the license agreement upon written notice to us under certain limited circumstances.
Ascletis Pharma
In September 2020, we announced that we entered into a research agreement with Gannex, a wholly owned company of Ascletis aiming at combination therapy of ASC41 (THR-beta agonist) and Aramchol (SCD 1 inhibitor) for the treatment NASH.
ASC41 is an oral thyroid hormone receptor beta (THR-beta) agonist which recently received IND approval from China’s NMPA to conduct clinical trials for Non-alcoholic Steatohepatitis (NASH) indication. In a Phase 1 study in 65 subjects with elevated low-density lipoprotein cholesterol (LDL-C) (> 110 mg/dL), a population characteristic of NAFLD, Ascletis reported that preliminary data suggested that ASC41 was safe and well tolerated up to a dose of 20 mg and in the multiple-ascending dose portion of the study, preliminary data suggest that after 14 days of once daily oral dosing, subjects demonstrate clinically meaningful and statistically significant reduction in LDL-C and triglycerides compared to placebo. We do not plan on proceeding with further research in combination therapy of ASC41 (THR-beta agonist) and Aramchol (SCD 1 inhibitor) for the treatment NASH.
MyBiotics
In November 2020, we announced that we entered into a research and development collaboration agreement with MyBiotics Pharma Ltd., or MyBiotics, to identify and optimize the selected microbiome repertoire associated with the response to Aramchol. The research focused on development of a standalone microbiome-based treatment for NASH and fibrosis. We do not plan on proceeding with further research.
Unipharm
On October 7, 2000, in connection with a certain share subscription agreement, we sent a letter to Unipharm Ltd., or Unipharm, pursuant to which we agreed to negotiate the grant of an exclusive license to Unipharm with respect to the use of patents within our first patent family covering the composition of matter of Aramchol within Israel on to-be-agreed upon terms and conditions. The letter stated that, if granted, such license would at all times be subject to our best interests, as determined in our sole discretion, and all approvals and proceedings required by agreement or by law. As of the date hereof, no such definitive agreement has been executed with regard to this matter and at this stage, we have no intention to pursue such an agreement. The letter is silent as to term, termination and whether or not it is binding.
Competition
The pharmaceutical industry is characterized by rapidly evolving technology, intense competition and a highly risky, costly and lengthy research and development process. Adequate protection of intellectual property, successful product development, adequate funding and retention of skilled, experienced and professional personnel are among the many factors critical to success in the pharmaceutical industry.
| 76 |
Along with our Phase 3 Aramchol study, other companies, including Madrigal Pharmaceuticals, Inc., Intercept Pharmaceuticals, Inventiva Pharma, Cirius Therapeutics and Novo Nordisk A/S, have molecules currently in Phase 3 clinical development. Additionally, there are a host of other potential competitors in earlier stages of clinical development relative to us for the treatment of NASH. Eli Lilly and Company, Pfizer, Novartis, Bristol-Myers Squibb, Novo Nordisk A/S, Merck, Viking Therapeutics, Inc., Metacrine, Inc., Poxel SA, Can-Fite BioPharma, 89bio, Inc., Sagimet Biosciences Inc. Terns, Inc., AstraZeneca and Hepanova Inc. and others.
In February 2019, Intercept Pharmaceuticals announced its Phase 3 results of their OCA drug for the treatment of liver fibrosis due to NASH and Intercept reported that it submitted an NDA to the FDA seeking accelerated approval of OCA for NASH and an MAA to the EMA. According to Intercept, in June 2020 it received a CRL from the FDA with respect to its NDA for OCA for liver fibrosis due to NASH and the CRL indicated that, based on the data the FDA had reviewed, the FDA has determined that the predicted benefit of OCA based on a surrogate histopathologic endpoint remains uncertain and does not sufficiently outweigh the potential risks to support accelerated approval for the treatment of patients with liver fibrosis due to NASH. Furthermore, according to Intecept, the FDA recommended that it submit additional post-interim analysis efficacy and safety data from its ongoing REGENERATE trial in support of potential accelerated approval and that the long-term outcomes phase of the trial should continue. According to Intercept, it is also in the process of generating a new data package from its REGENERATE study using the new liver biopsy consensus read methodology, and if the data supports accelerated approval, it plans to have a potential pre-NDA submission meeting with the FDA during the first half of 2022. Intercept reported that in December 2021, it withdrew its MAA as the established application timeline could not be extended any further to allow for submission of additional safety and efficacy data being generated from the REGENERATE study and the Committee for Medicinal Products for Human Use was not able to determine a positive benefit-risk based on previously submitted data.
Notwithstanding the foregoing, see “Item 3. Key Information—Risk Factors—Risks Related to Our Business, Industry and Regulatory Requirements—Our market is subject to intense competition. If we are unable to compete effectively, Aramchol or any other product candidate that we develop may be rendered noncompetitive or obsolete” and “Item 3. Key Information—Risk Factors—Risks Related to Our Business, Industry and Regulatory Requirements—We are developing Aramchol for the treatment of NASH, an indication for which there are no approved products, and there is significant uncertainty regarding the regulatory approval process. This makes it difficult to predict the timing and costs of the clinical development of Aramchol for the treatment of NASH.”
Intellectual Property and Patent Strategy
The proprietary nature of, and protection for, Aramchol or Aramchol meglumine or Amilo-5MER or any other product candidate and our discovery programs for new indications, processes and know-how are important to our business. We own patent rights to Aramchol and Aramchol meglumine or Amilo-5MER in various jurisdictions worldwide, including within and outside of Israel. We have sought patent protection in the United States and internationally for Aramchol or Amilo-5MER and our discovery programs, and any other inventions to which we have rights, where available and when appropriate. The term of U.S. Patent No. 7,501,403, covering the use of Aramchol for the treatment of fatty liver, has been extended due to patent term adjustments of 567 days, resulting in an effective expiration date of November 3, 2023. We have pending patent applications and have been granted patents directed to composition of matter of Aramchol meglumine as well as a wide range of other salts; a method for treating/inhibiting hepatic fibrosis and non-hepatic fibrosis associated or non-associated with non-alcoholic fatty acid liver disease; and a method of treating dysbiosis. In addition, we have pending patent applications for Aramchol meglumine and other salts, including a low dose composition for Aramchol meglumine and other salts. We have been granted patents for Aramchol meglumine and other salts which includes claims for the treatment of fatty liver in Europe and certain other countries while the composition of matter patent application is still pending in the U.S. and certain other countries (India and Brazil) and we have been granted a patent for Aramchol meglumine in the U.S. Our composition of matter patents which have claims directed to Aramchol meglumine and other salts; and low dose composition thereof, that have been granted - expire in 2034 and 2036, respectively (the low dose compositions US patent 2036 expiration is subject to terminal disclaimer (TD) over pending US patent application directed to Aramchol meglumine and other salts which will expire, if granted, at 2034), subject to appropriate maintenance, renewal, annuity or other governmental fees being paid. We have since been granted a patent in U.S. directed to a method for treating hepatic fibrosis non-associated with non-alcoholic fatty acid liver disease that will expire in 2037; and additional patent was granted in U.S., directed to a method of treating dysbiosis, that will expire in 2038 (including PTA). Also, a patent application was granted in Israel, directed to aramchol used in hepatic fibrosis treatment wherein administration regime is twice a day (BID).
| 77 |
We have a pending PCT international application, filed 2022, directed to composition of matter of scrambled variants of Amilo-5MER, and uses thereof (cancer and inflammatory disease treatment). “GALMED RESEARCH AND DEVELOPMENT LTD” is a co-applicant in this application with “YISSUM RESEARCH DEVELOPMENT COMPANY OF THE HEBREW UNIVERSITY OF JERUSALEM LTD”. The patent term for this patent family is due to expire about on March 15, 2042, not including any patent term extension.
We have an exclusive license to a US granted patent directed to Amilo-5MER and anti-inflammatory compositions thereof. The US Patent will be in full force and effect until July 15, 2035 subject to PTE of 84 day.
We also have a pending PCT international application, filed 2021, directed to peptides for treating acute respiratory distress syndrome. “GALMED RESEARCH AND DEVELOPMENT LTD” is a co-applicant in this PCT application with “YISSUM RESEARCH DEVELOPMENT COMPANY OF THE HEBREW UNIVERSITY OF JERUSALEM LTD. The patent term for this patent family is due to expire about on, 2041, not including any patent term extension.
Our policy is to pursue, maintain and defend patent rights, whether developed internally or licensed from third parties, and to protect the technology, inventions and improvements that are commercially important to the development of our business. We also rely on trade secrets that may be important to the development of our business.
Patent Portfolio for Aramchol and Aramchol Meglumine
The patent portfolio for Aramchol contains eight patent families including pending patent applications and granted patents directed to composition of matter, manufacturing methods and methods of use.
The first patent family discloses and claims additional FABACs with different conjugation moieties, as well as the use of these and the compounds disclosed in the first patent family above, including Aramchol, in the treatment of fatty liver, reduction of serum cholesterol and treatment of hyperglycemia and diabetes. This patent family includes a U.S. patent directed to the treatment of fatty liver a U.S. patent directed to reduction of serum cholesterol by administering additional forms of FABACs, and a U.S. patent (Continuation-in-Part) directed to the treatment of hyperglycemia and diabetes. This patent family also includes two European patents, one patent which was validated in Austria, Belgium, Cyprus, Denmark, Finland, France, Germany Ireland, Italy, Luxembourg, Monaco, Netherlands, Portugal, Spain, Sweden, Switzerland, Turkey and the United Kingdom, and the second patent which was granted in Belgium, Denmark, Finland, France, Germany, Greece, Ireland, Italy, Netherlands, Spain, Sweden, Switzerland, Turkey and the United Kingdom. The family also includes patents in Australia, Canada, China, Czech Republic, Azerbaijan, Belarus, Kyrgyzstan, Kazakhstan, Russian Federation, Indonesia, Japan, Korea, Israel, Mexico, New Zealand, Norway, Poland, Hungary and the Ukraine. A foreign patent application is granted in the Czech Republic. The non-extended patent term for this patent family expired on April 15, 2022, with the exception of the Israeli patent, which expired on April 17, 2021. The terms of the U.S. patents in this family have been extended due to patent term adjustments of 567 days for U.S. Patent 7,501,403, which is directed to the treatment of fatty liver, and 24 days for U.S. Patent 8,110,564, which is directed to reduction of serum cholesterol, and 356 days for U.S. Patent 8,975,246, which is directed to disorders associated with altered glucose metabolism or insulin action.
A second patent family directed to topical uses of FABAC compounds (anti-acne) was granted in Europe and maintained in Germany, France, Italy, the Netherlands and the United Kingdom. If appropriate and the appropriate maintenance, renewal, annuity or other governmental fees are paid, the non-extended term for this patent family is due to expire in August 2033, not including any patent term extension.
| 78 |
A third patent family discloses and claims second generation FABAC salt compounds include Aramchol meglumine. This patent family includes pending U.S., Brazil and India applications and granted patents in Europe (maintained in Albania, Austria, Bulgaria, Croatia, Cyprus, Czech Republic, Denmark, Estonia, Finland, Greece, Hungary, Iceland, Italy, Latvia, Lithuania, Macedonia, Malta, Norway, Poland, Portugal, Romania, San Marino, Serbia, Slovakia, Slovenia, Spain, Sweden, The Netherlands, Turkey, Belgium, France, Germany, Ireland, Luxembourg, Malta, Monaco, Switzerland and United Kingdom), China, Hong Kong, Macau, Canada, Israel, Korea and in Japan, as well as in Australia. If granted and the appropriate maintenance, renewal, annuity or other governmental fees are paid, the non-extended term for this patent family is due to expire in December 4, 2034, not including any patent term extension.
A fourth patent family having one U.S. patent application, discloses and claims compositions comprising low doses of the second generation FABAC compounds Aramchol meglumine and other salts which was granted by the USPTO. When the appropriate maintenance, renewal, annuity or other governmental fees are paid, the non-extended term for this patent family is due to expire in June 2036 (subject to terminal disclaimer (TD) over pending US patent application directed to Aramchol meglumine and other salts, which expires in December 2034).
A fifth family is directed to treatment for modulating gut microbiota and/or dysbiosis using Aramchol. This patent family includes a granted patents in Israel, and U.S.; allowed patent application in Mexico; and pending foreign patent applications in Brazil, Canada, China, Europe, Hong Kong and Japan. When the appropriate maintenance, renewal, annuity or other governmental fees are paid, the non-extended term for this patent family is due to expire in January 2036, not including any patent term extension. In U.S. the granted patent has a patent term adjustment (681 days), hence it is due to expire about December 2038.
A sixth patent family and eighth family, both having PCT international applications filed in 2017 and two pending US applications, are directed to uses of Aramchol and Aramchol meglumine for treating and inhibiting fibrosis. The two PCT applications entered National Phase in Australia, Brazil, Canada, China, Europe, Hong-Kong, Israel, Japan, Korea and Mexico. If granted and the appropriate maintenance, renewal, annuity or other governmental fees are paid, the non-extended term of this patent family is due to expire in November 2037, not including any patent term extension. Additionally, a U.S. patent application directed to a method for treating hepatic fibrosis non-associated with non-alcoholic fatty acid liver disease – was granted, and is due to expire in October 2037. In addition, a US continuation-in-part claiming priority to all of the above applications was filed in November 2018 and claims the treatment and inhibition of fibrosis by a regimen of 300 mg of Aramchol twice daily. The improved bio-availability of Aramchol is supported by the pharmacological model based on the preclinical and the ARREST data. Also, PCT application directed to treatment and inhibition of fibrosis by a regimen of 300 mg of Aramchol twice daily entered national phase in: Australia, Brazil, Canada, China, Europe, Israel and Mexico. The Israeli national phase directed to the treatment and inhibition of fibrosis by a regimen of 300 mg of Aramchol twice daily – was granted. If granted and the appropriate maintenance, renewal, annuity or other governmental fees are paid, the non-extended term of this patent family is due to expire in October 2039, not including any patent term extension.
A seventh patent family is directed to a combination therapy of FABAC and at least one thyroid hormone receptor agonist or thyroid hormone mimetic for treating fatty liver disease; and has a PCT application which entered National Phase in: Australia, Brazil, Canada, China, Europe, Hong Kong, Israel, India, Japan, Korea, Mexico and U.S. The patent term for this patent family is due to expire in September 3, 2038, not including any patent term extension.
Patent Portfolio for Amilo-5MER
The patent portfolio for Amilo-5MER contains three patent families including pending patent applications and granted patents directed to composition of matter and methods of use.
| 79 |
The first patent family is directed to Amilo-5MER and anti-inflammatory compositions thereof. GALMED RESEARCH AND DEVELOPMENT LTD has an exclusive license to the U.S. granted patent within this family, and this U.S. patent will expire at October 2035, including 84 days of patent term adjustment. GALMED RESEARCH AND DEVELOPMENT LTD has also exclusive licenses to the corresponding granted patents in Europe, Australia, Japan, China and India. The corresponding applications in Brazil, Korea and Canada are pending. The non-US patent term is due to expire on July 2035,.
The second patent family is directed to composition of matter of scrambled variants of Amilo-5MER, and uses thereof (cancer and inflammatory disease treatment). This family includes a pending PCT international application. “GALMED RESEARCH AND DEVELOPMENT LTD” is a co-applicant in this application with “YISSUM RESEARCH DEVELOPMENT COMPANY OF THE HEBREW UNIVERSITY OF JERUSALEM LTD”. The patent term for this patent family is due to expire about on March, 2042, not including any patent term extension.
The third patent family is directed to peptides for treating acute respiratory distress syndrome. This family includes a pending PCT international patent application. “GALMED RESEARCH AND DEVELOPMENT LTD” is a co-applicant in this PCT application with “YISSUM RESEARCH DEVELOPMENT COMPANY OF THE HEBREW UNIVERSITY OF JERUSALEM LTD”. The patent term for this patent family is due to expire about on August 2041, not including any patent term extension.
Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection of our current and other product candidates and the methods used to develop and manufacture them, as well as successfully defending these patents against third-party challenges. Our ability to stop third parties from making, using, selling, offering to sell or importing our products depends on the extent to which we have rights under valid and enforceable patents or trade secrets that cover these activities. We believe that our patents provide broad and comprehensive coverage of Aramchol and the Amilo-5MER and uses thereof. However, the patent positions of biopharmaceutical companies, such as ourselves, are generally uncertain and involve complex legal and factual questions. Our ability to maintain and solidify our proprietary position for the technology will depend on our success in obtaining effective claims and enforcing those claims once granted. There is no certainty that any of the Company’s pending patent applications will result in the issuance of any patents. The issued patents and those that may be issued in the future, may be challenged, narrowed, circumvented or found to be invalid or unenforceable, which could limit our ability to stop competitors from marketing related products or the length of term of patent protection that we may have for our products. In addition, our competitors may independently develop similar technologies or duplicate any technology developed by us, and the rights granted under any issued or future patents may not provide us with any meaningful competitive advantages against these competitors. Furthermore, because of the extensive time required for development, testing and regulatory review of a potential product, before any of our products can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of such patent. For more risks associated with the protection of our licensed intellectual property, see “Item 3. Key Information—Risk Factors—Risks Related to Our Intellectual Property.”
Trade Secrets
In addition to patents, we rely on trade secrets and know-how to develop and maintain our competitive position. Trade secrets and know-how can be difficult to protect. We seek to protect our proprietary processes, in part, by confidentiality agreements and invention assignment agreements with our employees, consultants, scientific advisors, contractors and commercial partners. These agreements are designed to protect our proprietary information. We also seek to preserve the integrity and confidentiality of our data, trade secrets and know-how by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, such agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors or others.
| 80 |
Seasonality
Our business and operations are generally not affected by seasonal fluctuations or factors.
Raw Materials and Suppliers
We believe that the raw materials that we require to manufacture Aramchol are readily available commodities commonly used in the pharmaceutical industry.
Manufacturing
We do not own or operate manufacturing facilities for the production of Aramchol or any other product candidate, nor do we have plans to develop our own manufacturing operations in the foreseeable future. We currently rely on third-party contract manufacturers for all of our required raw materials, API and finished product for our non-clinical research and clinical trials. We do not have long term agreements with any of these third parties. We also do not have any current contractual relationships for the manufacture of commercial supplies of Aramchol if it is approved. If Aramchol or any other product candidate are approved by any regulatory agency, we intend to enter into agreements with a third-party contract manufacturer or collaboration partner and one or more back-up manufacturers for the commercial production of those products. Development and commercial quantities of any products that we develop will need to be manufactured in facilities, and by processes, that comply with the requirements of the FDA and the regulatory agencies of other jurisdictions in which we are seeking approval. We currently employ internal resources to manage our manufacturing contractors. The relevant manufacturers of our drug substance and drug products for our current pre-clinical and clinical trials have advised us that they are compliant with both cGMP and, cGLP.
There can be no assurance that Aramchol, if approved, can be manufactured in sufficient commercial quantities, in compliance with regulatory requirements and at an acceptable cost. We and our contract manufacturers are, and will be, subject to extensive governmental regulation in connection with the manufacture of any pharmaceutical products or medical devices. We and our contract manufacturers must ensure that all of the processes, methods and equipment are compliant with cGMP and cGLP for drugs on an ongoing basis, as mandated by the FDA and other regulatory authorities, and conduct extensive audits of vendors, contract laboratories and suppliers.
Contract Research Organizations
We outsource certain clinical trial activities to CROs. Our clinical CROs comply with guidelines from the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, which attempt to harmonize the FDA, the EMA, and the Pharmaceuticals and Medical Devices Agency of Japan regulations and guidelines. We create and implement the drug development plans and manage the CROs according to the specific requirements of the drug candidate under development. To the extent clinical research is overseen by the CROs (or directly by us), compliance with certain federal regulations, including but not limited to 21 C.F.R. parts 50, 54, 56, 58 and 312, which pertain to, among other things, IRBs, informed consent, financial conflicts of interest by investigators, correct administration of treatment, follow up of adverse events, good laboratory practices and submitting IND applications, may be required.
Marketing, Sales and Commercialization
Given our stage of development, we do not have any internal sales, marketing or distribution infrastructure or capabilities. In the event we receive regulatory approval for any product candidate we intend, where appropriate, to pursue commercialization relationships, including strategic alliances and licensing, with pharmaceutical companies and other strategic partners, which are equipped to market and/or sell our product candidates through their well-developed sales, marketing and distribution organizations in order to gain access to global markets. In addition, we may out-license some or all of our worldwide patent rights to more than one party to achieve the fullest development, marketing and distribution of any products we develop. Over the longer term, we may consider ultimately building an internal marketing, sales and commercial infrastructure. See “Item 4. Information on the Company—Business Overview—Strategic Collaborations, Research Arrangements and other Material Agreements—Samil Pharm Co.” for information regarding the license agreement we entered with Samil for the commercialization of Aramchol (with an option to manufacture) for the treatment of fatty liver indications including NASH, in the Republic of Korea.
| 81 |
Environmental Matters
We, our agents and our service providers, including our manufacturers, may be subject to various environmental, health and safety laws and regulations, including those governing air emissions, water and wastewater discharges, noise emissions, the use, management and disposal of hazardous, radioactive and biological materials and wastes and the cleanup of contaminated sites. We believe that our business, operations and facilities, including, to our knowledge, those of our agents and service providers, are being operated in compliance in all material respects with applicable environmental and health and safety laws and regulations. All information with respect to any chemical substance is filed and stored as a Material Safety Data Sheet, as required by applicable environmental regulations. Based on information currently available to us, we do not expect environmental costs and contingencies to have a material adverse effect on us. However, significant expenditures could be required in the future if we, our agents or our service providers are required to comply with new or more stringent environmental or health and safety laws, regulations or requirements.
Government Regulation and Product Approval
Governmental authorities in the United States and in other countries extensively regulate, among other things, the research, development, testing, manufacture, labeling, packaging, promotion, storage, advertising, distribution, marketing and export and import of products such as those we are developing. Aramchol or any other product candidate must be approved by the FDA through the NDA process before they may be legally marketed in the United States and by the Committee on Human Medicinal Products, or CHMP, via the EMA and European Commission through the MAA process before they may be legally marketed in Europe, or MHRA through its authorization procedures before they may be legally marketed in the UK. Aramchol or any other product candidate will be subject to similar requirements in other countries prior to marketing in those countries. The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
United States Government Regulation
NDA Approval Processes
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or the FDCA, and implementing regulations and guidance documents. Failure to comply with the applicable U.S. requirements at any time during the product development process or approval process, or after approval, may subject an applicant to administrative or judicial sanctions, any of which could have a material adverse effect on us. These sanctions could include refusal to approve pending applications, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters, product seizures, total or partial suspension of production or distribution, injunctions, fines, disgorgement, and civil or criminal penalties.
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
| ● | completion of pre-clinical laboratory tests, animal studies and formulation studies conducted according to GLPs, or other applicable regulations; |
| 82 |
| ● | submission to the FDA of an IND application, which must become effective before human clinical trials may begin; | |
| ● | performance of adequate and well-controlled human clinical trials according to GCPs, to establish the safety and efficacy of the proposed drug for its intended use; | |
| ● | submission to the FDA of an NDA; | |
| ● | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with cGMPs to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; | |
| ● | satisfactory completion of FDA inspections of clinical sites and GLP toxicology studies; and | |
| ● | FDA review and approval of the NDA. |
The testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for Aramchol, Amilo-5MER or any other product candidate will be granted on a timely basis, if at all.
Once a product candidate is identified for development, it enters the pre-clinical testing stage. Pre-clinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies. An IND sponsor must submit the results of the pre-clinical tests, together with manufacturing information and analytical data, to the FDA as part of the IND. Some pre-clinical testing may continue after the IND is submitted. In addition to including the results of the pre-clinical studies, the IND will also include a clinical trial protocol detailing, among other things, the objectives of the clinical trial, the parameters to be used in monitoring safety and, depending on the phase of the study, the effectiveness criteria to be evaluated. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, places the IND on clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can begin. A clinical hold may occur at any time during the life of an IND, due to safety concerns or non-compliance, and may affect one or more specific studies or all studies conducted under the IND.
All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with the FDA’s GCP regulations. These regulations include the requirement that all research subjects provide informed consent. Further, an IRB must review and approve the plan for any clinical trial, including the informed consent document, before it commences at any institution. An IRB considers, among other things, whether the risks to individuals participating in the trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the investigator brochure and other information about the trial distributed by the sponsor and the consent form that must be provided to each trial subject or his or her legal representative and must monitor the study until completed. All clinical trials must be conducted under protocols detailing the objectives of the trial, dosing procedures, research subject inclusion and exclusion criteria and the safety and effectiveness criteria to be evaluated. Each protocol must be submitted to the FDA as part of the IND, and progress reports detailing the status of the clinical trials must be submitted to the FDA annually. Sponsors must also report within set timeframes to FDA serious and unexpected adverse reactions, any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigation brochure, or any findings from other studies or animal or in-vitro testing that suggest a significant risk in humans exposed to the drug. Sponsors must also report to FDA certain amendments to the protocol and other essential information concerning the IND that does not fall within the scope of other required reports.
| 83 |
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| ● | Phase 1. The drug is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and elimination. In the case of some products for severe or life-threatening diseases, such as cancer, especially when the product may be inherently too toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients. | |
| ● | Phase 2. Clinical trials are performed on a limited patient population intended to identify possible adverse effects and risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. | |
| ● | Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical study sites. Phase 3 clinical trials are conducted to provide sufficient data for the statistically valid evidence of safety and efficacy. | |
| ● | Phase 4. The FDA may require that the sponsor conduct additional clinical trials following new drug approval. The purpose of these trials, known as Phase 4 studies, is to monitor long-term risks and benefits, study different dosage levels or evaluate safety and effectiveness. In recent years, the FDA has increased its reliance on these trials. Phase 4 studies usually involve thousands of participants. Phase 4 studies also may be initiated by the company sponsoring the new drug to gain broader market value for an approved drug. |
Human clinical trials are inherently uncertain and Phase 1, Phase 2, Phase 3 and Phase 4 testing may not be successfully completed. The FDA or the sponsor may suspend a clinical trial at any time for a variety of reasons, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
During the development of a new drug, sponsors are given opportunities to meet with the FDA at certain points. These points are typically prior to the submission of an IND, at the end of Phase 2 and before an NDA is submitted. Meetings at other times may also be requested. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date and for the FDA to provide advice on the next phase of development. Sponsors typically use the meeting at the end of Phase 2 to discuss their Phase 2 clinical results and present their plans for the pivotal Phase 3 clinical trial that they believe will support the approval of the NDA.
Concurrent with clinical trials, sponsors usually complete any remaining animal safety studies and also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing commercial quantities of the product in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug and the manufacturer must develop methods for testing the quality, purity and potency of the drug. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its proposed shelf-life.
The results of product development, pre-clinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests and other control mechanisms, proposed labeling and other relevant information are submitted to the FDA as part of an NDA requesting approval to market the product for one or more specified indications. The submission of an NDA is subject to the payment of an application fee, but a waiver of such fees may be obtained under specified circumstances. We will seek a waiver of these fees as a small business submitting its first human drug application to the FDA. If the waiver is granted it would not extend to establishment or product fees. The FDA reviews all NDAs submitted to ensure that they are sufficiently complete for substantive review before it accepts them for filing. It may request additional information rather than accept an NDA for filing. In this event, the NDA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing.
| 84 |
Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA may refuse to approve an NDA if the applicable statutory and regulatory criteria are not satisfied or may require additional clinical or other data. Even if such data are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. The FDA reviews an NDA to determine, among other things, whether a product is safe and effective for its intended use and whether its manufacturing is cGMP-compliant. The FDA may refer the NDA to an advisory committee for review and recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving an NDA, the FDA will typically inspect the facility or facilities where the product is manufactured and tested. The FDA will also inspect selected clinical sites that participated in the clinical studies and may inspect the testing facilities that performed the GLP toxicology studies cited in the NDA.
Expedited Review and Approval
The FDA has various specific programs, including Fast Track, Breakthrough Therapy, Priority Review, and Accelerated Approval, which, in different ways, are each intended to expedite the process for reviewing and approving drugs. Even if a drug qualifies for one or more of these programs, the FDA may later decide that the drug no longer meets the conditions for qualification or that the time period for FDA review or approval will be shortened. Generally, drugs that are eligible for these programs are those for serious or life-threatening conditions, those with the potential to address unmet medical needs and those that offer meaningful benefits over existing treatments. For example, Fast Track is a process designed to facilitate the development and expedite the review of drugs to treat serious or life-threatening diseases or conditions and fill unmet medical needs, and Breakthrough Therapy designation is designed to expedite the development and review of drugs that are intended to treat a serious condition where preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s). Priority review is designed to give drugs that offer major advances in treatment or provide a treatment where no adequate therapy exists an initial review within six months as compared to a standard review time of ten months. Although Fast Track, Breakthrough Therapy designation and priority review do not affect the standards for approval, the FDA will attempt to facilitate early and frequent meetings with a sponsor of a Fast Track or Breakthrough Therapy designated drug and expedite review of the application for a drug designated for priority review. The FDA will also provide Breakthrough Therapy designated drugs intensive guidance on an efficient drug development program and provide these drug developers with an organizational commitment from the FDA involving senior managers. Since sponsors can design clinical trials in a number of ways, in providing its guidance for drugs designated as breakthrough therapies, the FDA will seek to ensure that the sponsor of the product designated as a breakthrough therapy receives timely advice and interactive communications in order to help the sponsor design and conduct a development program as efficiently as possible. During these interactions, the FDA may suggest, or a sponsor can propose, alternative clinical trial designs (e.g., adaptive designs, an enrichment strategy, use of historical controls) that may result in smaller trials or more efficient trials that require less time to complete. Such trial designs could also help minimize the number of patients exposed to a potentially less efficacious treatment (i.e., the control group treated with available therapy). On September 23, 2014, the FDA granted Fast Track designation status to Aramchol for the treatment of patients who are overweight or obese and have pre diabetes or type II diabetes mellitus with NASH.
Accelerated Approval, which is described in 21 C.F.R. § 314.500 et seq., provides for approval of a new drug that is intended to treat a serious or life-threatening disease or condition and that fills an unmet medical need based on a surrogate endpoint. A surrogate endpoint is a laboratory measurement or physical sign used as an indirect or substitute measurement representing a clinically meaningful outcome. To be used in accelerated approval, a surrogate endpoint must be “reasonably likely, based on epidemiologic, therapeutic, pathophysiologic, or other evidence to predict benefit on irreversible morbidity or mortality.” The term “reasonably likely” implies that some uncertainty remains about the relationship of the surrogate to the clinical benefit to the patient. Therefore, accelerated approval is typically contingent on a sponsor’s agreement to conduct additional post-approval studies to verify and describe the drug’s clinical benefit. Accelerated Approval does not change the standards for approval, but by allowing a demonstration of efficacy based on a surrogate endpoint may expedite the approval process.
| 85 |
FDA Guidance
In December 2018, the FDA issued “Noncirrhotic Nonalcoholic Steatohepatitis with Liver Fibrosis: Developing Drugs for Treatment”, or “the December Guidance”. The December Guidance, though nonbinding on the FDA or us, is intended to assist sponsors in the clinical development of drugs for the treatment of noncirrhotic NASH with liver fibrosis, describes the FDA’s current thinking regarding the necessary components of a drug development program for noncirrhotic NASH with liver fibrosis and identifies knowledge gaps that represent important challenges in the development of drugs for the indication. According to the FDA, the ultimate goal of NASH treatment is to slow the progress of, halt, or reverse disease progression and improve clinical outcomes (i.e., prevent progression to cirrhosis and cirrhosis complications, reduce the need for liver transplantation, and improve survival). Because of the slow progression of NASH and the time required to conduct a trial that would evaluate clinical endpoints such as progression to cirrhosis or survival, the FDA recommends sponsors consider the following liver histological improvements as endpoints reasonably likely to predict clinical benefit to support accelerated approval under the regulations:
| ● | Resolution of steatohepatitis on overall histopathological reading and no worsening of liver fibrosis on NASH CRN fibrosis score. Resolution of steatohepatitis is defined as absent fatty liver disease or isolated or simple steatosis without steatohepatitis and a NAS score of 0–1 for inflammation, 0 for ballooning, and any value for steatosis; or | |
| ● | Improvement in liver fibrosis greater than or equal to one stage (NASH CRN fibrosis score) and no worsening of steatohepatitis (defined as no increase in NAS for ballooning, inflammation, or steatosis) |
Further, according to the FDA, for NASH drugs approved on the basis of liver histology under the accelerated approval pathway, randomized, double-blind, placebo-controlled clinical trials designed to describe and verify the drug’s clinical benefit should be underway at the time of submission of the marketing application. Clinical benefit can be verified by demonstrating superiority to placebo in delaying disease progression measured by a composite endpoint.
The EMA also issued a reflection paper to provide guidance on drug development in the field of NASH. However, the EMA indicated, among other things, that both resolution of NASH without worsening of fibrosis and improvement in fibrosis without worsening of NASH would both be required as intermediate endpoints for demonstrating statistical significance for stage 2 and 3 fibrosis.
See also “Item 3. Key Information—Risk Factors—Risk Factors—Risks Related to Our Business, Industry and Regulatory Requirements - We are developing Aramchol for the treatment of NASH, an indication for which there are no approved products, and there is significant uncertainty regarding the regulatory approval process. This makes it difficult to predict the timing and costs of the clinical development of Aramchol for the treatment of NASH.”
Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of FDA approval of the use of Aramchol or any other product candidate, U.S. patents may be eligible for limited patent term extension under the Hatch-Waxman Act. The Hatch-Waxman Act permits a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND, and the submission date of an NDA, plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension and the application for extension must be made prior to expiration of the patent. The USPTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we intend to apply for restorations of patent term for some of our currently owned patents to add patent life beyond their current expiration date, depending on the expected length of clinical trials and other factors involved in the submission of the relevant NDA.
| 86 |
Market exclusivity provisions under the FDCA can also delay the submission or the approval of certain applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an approved NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, for new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA; however, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the pre-clinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
Post-approval Requirements
Once an approval is granted, the FDA, European authorities and other regulatory authorities may withdraw the approval if compliance with regulatory requirements is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further regulatory authority review and approval. Some of these modifications, especially adding indications, would likely require additional clinical studies. In addition, the FDA may require testing and surveillance programs to monitor the effect of approved products that have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs.
Any drug product manufactured or distributed by us pursuant to FDA approvals are subject to continuing regulation by the FDA, including, among other things record-keeping requirements; cGMPs; reporting of adverse experiences with the drug; providing the FDA with updated safety and efficacy information; drug sampling and distribution requirements; notifying the FDA and gaining its approval of specified manufacturing or labeling changes; and complying with FDA promotion and advertising requirements.
Drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and some state agencies for compliance with cGMP and other laws.
We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of Aramchol. Future FDA and state inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct.
From time to time, legislation is drafted, introduced and passed in Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. In addition, FDA regulations and guidance are often revised or reinterpreted by the agency in ways that may significantly affect our business and our products. It is impossible to predict whether legislative changes will be enacted, or FDA regulations, guidance or interpretations changed or what the impact of such changes, if any, may be. In particular, it is unknown whether any of the provisions of the 2016 21st Century Cures Act that are intended to accelerate drug approval will result in any change in the current approval pathway for Aramchol.
| 87 |
Pursuant to the Affordable Care Act (discussed in greater detail below), the Centers for Medicare & Medicaid Services (CMS) is required to collect and publish information reported by applicable manufacturers about payments and other transfers of value manufacturers have made to physicians and teaching hospitals. Such a law, when applicable to our products, could increase the company’s regulatory liability through the imposition of additional reporting and regulatory requirements. There are also an increasing number of state laws that require manufacturers to make similar reports to states on pricing and marketing information.
Reimbursement
We face uncertainties over the pricing of pharmaceutical products. Sales of Aramchol or any other product candidate will depend, in part, on the extent to which the costs of Aramchol or any other product candidate will be covered by third-party payors, such as federal health programs, commercial insurance and managed care organizations. These third-party payors are increasingly challenging the prices charged for medical products and services. Additionally, the containment of healthcare costs has become a priority of federal and state governments and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures, foreign governments and third party payors have shown significant interest in implementing cost-containment programs, including price controls, pricing transparency disclosure obligations, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. If these third-party payors do not consider Aramchol or any other product candidate to be cost-effective compared to other therapies, they may not cover Aramchol or any other product candidate after approved as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell Aramchol or any other product candidate on a profitable basis.
The Medicare Modernization Act imposed new requirements for the distribution and pricing of prescription drugs for Medicare beneficiaries under Part D. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities that provide coverage of outpatient prescription drugs. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. The Centers for Medicare & Medicaid Services published a final rule in 2014 implementing the Medicare Modernization Act. Contrary to the proposed rule, which would have enabled Part D plans to offer fewer drugs, the final rule maintained the existing six protected classes of drug categories, but stated that some of the proposals not included in the final rule could still be finalized in the future, which would impact payor formulary and coverage decisions.
The American Recovery and Reinvestment Act of 2009 provides funding for the federal government to compare the effectiveness of different treatments for the same illness. A plan for the research will be developed by the Department of Health and Human Services, the Agency for Healthcare Research and Quality and the National Institutes for Health, and periodic reports on the status of the research and related expenditures will be made to Congress. Although the results of the comparative effectiveness studies are not intended to mandate coverage policies for public or private payors, it is not clear what effect, if any, the research will have on the sales of any product, if any such product or the condition that it is intended to treat is the subject of a study. It is also possible that comparative effectiveness research demonstrating benefits in a competitor’s product could adversely affect the sales of Aramchol or any other product candidate. If third-party payors do not consider Aramchol or any other product candidate to be cost-effective compared to other available therapies, they may not cover Aramchol or any other product candidate as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell Aramchol or any other product candidate on a profitable basis.
| 88 |
The Affordable Care Act, enacted in March 2010, has had a significant impact on the health care industry. Some of the key changes made to date pursuant to the Affordable Care Act include an expansion of coverage for the uninsured, the creation of insurance marketplaces and increased protection of insureds with new benefits, rights and protections. With regard to pharmaceutical products, among other things, the Affordable Care Act made major changes to the Medicare prescription drug program, which helped reduce drug costs for seniors and increased rebates and other costs for the pharmaceutical industry.
There have been judicial and congressional challenges to the Affordable Care Act. In December 2017, Congress passed and then the President Trump signed into law tax reform legislation that made significant changes to the Affordable Care Act including the repeal of the “individual mandate” that was in place to strongly encourage broad participation in the health insurance markets. On December 14, 2018, a federal district court in Texas ruled that the PPACA is unconstitutional as a result of the Tax Cuts and Jobs Act, the federal income tax reform legislation previously passed by Congress and signed by President Trump on December 22, 2017, that eliminated the individual mandate portion of the PPACA. The case, Texas, et al, v. United States of America, et al., (N.D. Texas), is an outlier, but in 2019, the Fifth Circuit Court of Appeals subsequently upheld the lower court decision which was then appealed to the United States Supreme Court. The U.S. Supreme Court declined to hear the appeal on an expedited basis and so no decision is expected until sometime in 2021 before the end of the Supreme Court’s current term. We are not able to state with any certainty what will be impact of this court decision on our business pending further court action and possible appeals. Given these changes and other statements of political leaders, we cannot predict the ultimate impact on the Affordable Care Act and the subsequent effect on the pharmaceutical industry at this time. In November 2020, Joseph Biden was elected President and, in January 2021, the Democratic Party obtained control of the Senate. As a result of these electoral developments, it is unlikely that continued legislative efforts will be pursued to repeal PPACA. Instead, it is possible that executive and regulatory initiatives, as well as legislation, will be pursued to enhance or reform PPACA. We are not able to state with certainty what the impact of potential legislation will be on our business.
In addition, in some non-U.S. jurisdictions, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the EU provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for Aramchol, Amilo-5MER or any other product candidate. Historically, products launched in the EU do not follow price structures of the United States and generally tend to be significantly lower.
Healthcare Fraud and Abuse Laws
In the U.S., the research, development, testing, manufacturing, handling, storage, distribution, sale and promotion of drug products and medical devices are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including the Centers for Medicare & Medicaid Services, other divisions of the U.S. Department of Health and Human Services (e.g., the Office of Inspector General), the U.S. Department of Justice, state Attorneys General, and other state and local government agencies. For example, sales, marketing and scientific/educational grant programs must comply with the fraud and abuse provisions applicable to pharmaceutical manufacturers, including the federal “Anti-Kickback Statute”, the Civil Monetary Penalty Statute, the Stark Law, the federal False Claims Act, as amended, state and federal “Physician Payment Sunshine Act” laws and regulations, the privacy regulations promulgated under the Health Insurance Portability and Accountability Act, or HIPAA, and similar state laws. Pricing and rebate programs must comply with the Medicaid Drug Rebate Program requirements of the Omnibus Budget Reconciliation Act of 1990, as amended, and the Veterans Health Care Act of 1992, as amended. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. All of these activities are also potentially subject to federal and state consumer protection and unfair competition laws. Some of these health care laws include:
| 89 |
The Anti- Kickback Statute makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf) to knowingly and willfully solicit, receive, offer, or pay any remuneration that is intended to induce the referral of business, including the purchase, order, or prescription of a particular drug, for which payment may be made under a federal healthcare program, such as Medicare or Medicaid.
The federal False Claims Act prohibits anyone from knowingly presenting, conspiring to present, making a false statement in order to present, or causing to be presented, for payment to federal programs (including Medicare and Medicaid) claims for items or services, including drugs, that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services. This law also prohibits anyone from knowingly underpaying an obligation owed to a federal program. Increasingly, U.S. federal agencies are requiring nonmonetary remedial measures, such as corporate integrity agreements in False Claims Act settlements. The U.S. Department of Justice announced in 2016 its intent to follow the “Yates Memo,” taking a far more aggressive approach in pursuing individuals as False Claims Act defendants in addition to the corporations.
The Physician Payment Sunshine Act, enacted in 2010 as part of the Affordable Care Act, requires certain manufacturers of pharmaceuticals and medical devices to annually report certain payments and other transfers of value to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as well as investment interests held by physicians and their immediate family members. Effective January 1, 2022, covered manufacturers will also be required to report on payments and other transfers of value to physician assistants, nurse practitioners or clinical nurse specialists, anesthesiologist assistants, certified registered nurse anesthetists, and certified nurse-midwives during the previous year. In recent years, several states in the United States have also enacted legislation requiring pharmaceutical companies to file periodic reports with the state, make periodic public disclosures on sales, marketing, pricing, clinical trials and other activities, and/or register their sales representatives, as well as establish marketing compliance programs. These laws may affect our sales, marketing, and other promotional activities by imposing administrative and compliance burdens on us. Failure to meet these requirements, to the extent they are applicable to our activities, could also result in a variety of governmental sanctions that could have a material adverse effect on our business.
If our operations are found to be in violation of any of the foregoing or other applicable health care laws and regulations, we may be subject to penalties, including significant administrative, civil and criminal penalties, monetary damages, disgorgement, imprisonment, the curtailment or restructuring of our operations, loss of eligibility to obtain approvals from the FDA, or exclusion from participation in government contracting, healthcare reimbursement or other government programs, including Medicare and Medicaid.
European Economic Area
In addition to approval in the United States, we currently intend to seek regulatory approval of Aramchol in the EU. As such, a summary of the EU regulatory processes follows below.
A medicinal product may only be placed on the market in the European Economic Area, or the EEA, composed of the 27 EU member states of the EU, plus Norway, Iceland and Lichtenstein, when a marketing authorization has been issued by the competent authority of the respective member state pursuant to member states’ law based on Directive 2001/83/EC, or an authorization has been granted under the centralized procedure in accordance with Regulation (EC) No. 726/2004 or its predecessor, Regulation 2309/93. There are essentially three community procedures created under prevailing European pharmaceutical legislation that, if successfully completed, allow an applicant to place a medicinal product on the market in the respective EU or EEA member states.
| 90 |
The withdrawal of the United Kingdom (UK) from the EU took effect on January 1, 2021, and there are 27 member states remaining in the EU. As of January 1, 2021, the UK is a “third country” with regard to the EU (subject to the terms of the EU UK Trade Agreement) and EU law ceased to apply directly in the UK. However, the UK has retained the EU medicines regulatory regime with certain modifications as standalone UK legislation. Therefore, the UK regulatory regime is currently similar to EU regulations, but under new legislation, the Medicines and Medical Devices Act 2021, the UK may adopt changed regulations that may diverge from the EU legislative regime for medicines and their research, development and commercialization. In order to market a medicinal product in the UK, a license or marketing authorization must be obtained from the UK Medicines and Healthcare Products Regulatory Agency (MHRA). The UK legislation includes multiple assessment routes for applications for medicinal products, including a 150-day national assessment or a rolling review application. Further, and for a transitional period until 31 December 2022, the MHRA may rely on a decision taken by the European Commission on the approval of a new marketing authorization in the centralized procedure. In addition, the MHRA has the power to have regard to marketing authorizations approved in EU member states through the decentralized and mutual recognition procedures.
Centralized Procedure
Regulation 726/2004/EC governs the centralized procedure when a marketing authorization is granted by the European Commission, acting in its capacity as the European Licensing Authority on the advice of the EMA. That authorization is valid throughout the entire community and directly or (as to Norway, Iceland and Liechtenstein) indirectly allows the applicant to place the product on the market in all member states of the EEA. The EMA is the administrative body responsible for coordinating the existing scientific resources available in the member states for evaluation, supervision and pharmacovigilance of medicinal products. Certain medicinal products, as described in the Annex to Regulation 726/2004, must be authorized centrally. These are products that are developed by means of certain biotechnological processes in accordance with Paragraph 1 to the Annex to the Regulation. Medicinal products for human use containing a new active substance for which the therapeutic indication is the treatment of acquired immune deficiency syndrome, or AIDS, cancer, neurodegenerative disorder or diabetes, autoimmune diseases and other immune dysfunctions and viral diseases must also be authorized centrally. Finally, all medicinal products that are designated as orphan medicinal products pursuant to Regulation 141/2000 and Advanced Therapy Medicinal Products (ATMP) according to Reg. (EC) No. 1394/2007 and medicinal products for veterinary use that are used primarily as performance enhancers must be authorized under the centralized procedure. An applicant may also opt for assessment through the centralized procedure if the medicinal product contains a new active substance which was not authorized in the EU when Reg. (EC) No. 726/2004 entered into force, or if the applicant can show that the medicinal product constitutes a significant therapeutic, scientific or technical innovation or that the granting of authorization centrally is in the interests of patients or animal health at the community level. For each application submitted to the EMA for scientific assessment, the EMA is required to ensure that the opinion of the Committee for Medicinal Products for Human Use, or CHMP, is given within 210 days after receipt of a valid application. This 210 days period does not include the time that the applicant needs to answer any questions raised during the application procedure, the so-called ‘clock stop’ period. If the opinion is positive, the EMA is required to send the opinion to the European Commission, which is responsible for preparing the draft decision granting a marketing authorization. This draft decision may differ from the CHMP opinion, stating reasons for diverging from the CHMP opinion. The draft decision is sent to the applicant and the member states, after which the European Commission takes a final decision. If the initial opinion of the CHMP is negative, the applicant is afforded an opportunity to seek a re-examination of the opinion. The CHMP is required to re-examine its opinion within 60 days following receipt of the request by the applicant. All CHMP refusals and the reasons for refusal are made public on the EMA website. Without a centralized marketing authorization it is prohibited to place a medicinal product that must be authorized centrally on the market in the EU. Once a centralized marketing authorization has been granted by the European Commission, it is valid in all EEA States for 5 years on a renewable basis.
| 91 |
Mutual Recognition and Decentralized Procedures
With the exception of products that are authorized centrally, the competent authorities of the member states are responsible for granting marketing authorizations for medicinal products placed on their national markets. If the applicant for a marketing authorization intends to market the same medicinal product in more than one member state, the applicant may seek an authorization progressively in the community under the mutual recognition or decentralized procedure. Mutual recognition procedure, or MRP is used if the medicinal product has already been authorized in a member state. In this case, the holder of this marketing authorization requests the member state where the authorization has been granted to act as reference member state by preparing an updated assessment report that is then used to facilitate mutual recognition of the existing authorization in the other member states in which approval is sought (the so-called concerned member state(s)). The reference member state must prepare an updated assessment report within 90 days of receipt of a valid application. This report together with the approved Summary of Product Characteristics, the SmPC (which sets out the conditions of use of the product), and a labeling and package leaflet are sent to the concerned member states for their consideration. The concerned member states are required to approve the assessment report, the SmPC and the labeling and package leaflet within 90 days of receipt of these documents. The total procedural time of the MRP is 180 days.
The decentralized procedure, or DCP, is used in cases where the medicinal product has not received a marketing authorization in the EU at the time of application. The applicant requests a member state of its choice to act as reference member state to prepare an assessment report that is then used to facilitate agreement with the concerned member states and the grant of a national marketing authorization in all of these member states. In this procedure, the reference member state must prepare, for consideration by the concerned member states, the draft assessment report, a draft SmPC and a draft of the labeling and package leaflet within 120 days after receipt of a valid application. As in the case of mutual recognition, the concerned member states are required to approve these documents within 90 days of their receipt, i.e. the total time of the DCP is 210 days.
For both MRP and DCP, if a concerned member state objects to the grant of a marketing authorization on the grounds of a potential serious risk to public health, it may raise a reasoned objection with the reference member state. The points of disagreement are in the first instance referred to the Co-ordination Group on MRP and DCP to reach an agreement within 60 days of the communication of the points of disagreement. If member states fail to reach an agreement, then the matter is referred to the EMA and CHMP for arbitration. The CHMP is required to deliver a reasoned opinion within 60 days of the date on which the matter is referred. The scientific opinion adopted by the CHMP forms the basis for a binding European Commission decision.
Irrespective of whether the medicinal product is assessed centrally, de-centrally or through a process of mutual recognition, the medicinal product must be manufactured in accordance with the principles of GMP as set out in Directive 2001/83/EC and Directives 2017/1569 and 2017/1572/EU that have replaced Directive 2003/94/EC. Directive 2003/94/EC will still be applicable to clinical trials conducted in accordance with the former regime under transitional provisions.
Directive 2017/1572/EU and Volume 4 of the rules governing medicinal products in the European Union govern GMP in the EU. Moreover, EU law requires the clinical results in support of clinical safety and efficacy based upon clinical trials conducted in the EU to be in compliance with the requirements of Regulation (EU) 536/2014 on clinical trials with medicinal products for human use and Implementing Regulation 556/2017 on GCP-inspections which implement good clinical practice in the conduct of clinical trials on medicinal products for human use. Clinical trials conducted outside the European community and used to support applications for marketing within the EU must have been conducted in a way consistent with the principles set out in Regulation (EU) 536/2017. The conduct of a clinical trial in the EU requires, pursuant to Regulation (E) 536/2017, authorization by the relevant national competent authority where a trial takes place, and subject to the relevant national law, an ethics committee to have issued a favorable opinion in relation to the arrangements for the trial. It also requires that the sponsor of the trial, or a person authorized to act on his behalf in relation to the trial, be established in the community. In comparison to the former regime, Regulation (EU) 536/2014 further harmonizes EU law on clinical trials and to some extent facilitates clinical trials conducted in more than one EU Member State. Under Regulation (EU) 536/2014, trial sponsors submit their application for trial approval via an EU Portal. The approvals will still have to be granted by the competent authorities of the EU Member States where a trial takes place, however, the procedure for approval is conducted in a coordinated manner among the concerned EU Member States as provided under Regulation (EU) 536/2014. While the process for the application and granting of the approvals was streamlined, it is still a complex process that can significantly delay the start of a multinational clinical trial.
| 92 |
The UK has not adopted Regulation (EU) 536/2014 with respect to clinical trials in the UK. Instead, the Medicines for Human Use (Clinical Trials) Regulations 2004 applies with respect to clinical trials in the UK. The UK has adopted new legislation, the Medicines and Medical Devices Act 2021 and has issued a consultation with respect to changes to clinical trial legislation to be made under this Act.
National Procedure
This procedure is available for medicinal products that do not fall within the scope of mandatory centralized authorization. Specific procedures and timelines differ between member states, but the duration of the procedure without clock-stop time is generally 210 days and based on a risk/efficacy assessment by the competent authority of the member state concerned, followed by determination of SmPC, package leaflet and label text/layout and subsequently grant of the marketing authorization. Marketing authorizations granted on this basis are not mutually recognized by other member states, but the national marketing authorization can later be used in an MRP to obtain marketing authorizations in other member states.
There are various types of applications for marketing authorizations:
| ● | Full Applications. A full application is one that is made under any of the community procedures described above and that “stands alone” in the sense that it contains all of the particulars and information required by Article 8(3) of Directive 2001/83 (as amended) to allow the competent authority to assess the quality, safety and efficacy of the product and in particular the balance between benefit and risk. Article 8(3)(l) in particular refers to the need to present the results of the applicant’s research on (i) pharmaceutical (physical-chemical, biological or microbiological) tests, (ii) pre-clinical (toxicological and pharmacological) studies and (iii) clinical trials in humans. The nature of these tests, studies and trials is explained in more detail in Annex I to Directive 2001/83/EC. Full applications would be required for products containing new active substances not previously approved by the competent authority, but may also be made for other products. | |
| ● | Abridged Applications. Article 10 of Directive 2001/83/EC contains exemptions from the requirement that the applicant has to provide the results of its own pre-clinical and clinical research. There are three regulatory routes for an applicant to seek an exemption from providing such results, namely (i) cross-referral to an innovator’s results without consent of the innovator, (ii) well established use according to published literature and (iii) consent to refer to an existing dossier of research results filed by a previous applicant. |
Cross-referral to Innovator’s Data
Articles 10(1) and 10(2)(b) of Directive 2001/83/EC provide the legal basis for an applicant to seek a marketing authorization on the basis that its product is a generic medicinal product (a copy) of a reference medicinal product that has already been authorized, in accordance with community provisions. A reference product is, in principle, an original product granted an authorization on the basis of a full dossier of particulars and information. This is the main exemption used by generic manufacturers for obtaining a marketing authorization for a copy product. The generic applicant is not required to provide the results of pre-clinical studies and of clinical trials if its product meets the definition of a generic medicinal product and the applicable regulatory results protection period for the results submitted by the innovator has expired. A generic medicinal product is defined as a medicinal product:
| ● | having the same qualitative and quantitative composition in active substance as the reference medicinal product; |
| 93 |
| ● | having the same pharmaceutical form as the reference medicinal product; and | |
| ● | whose bioequivalence with the reference medicinal product has been demonstrated by appropriate bioavailability studies. |
Applications in respect of a generic medicinal product cannot be made before the expiry of the protection period. Where the reference product was granted a national marketing authorization pursuant to an application made before October 30, 2005, the protection period is either six years or 10 years, depending upon the election of the particular member state concerned. Where the reference product was granted a marketing authorization centrally, pursuant to an application made before November 20, 2005, the protection period is 10 years. For applications made after these dates, Regulation 726/2004 and amendments to Directive 2001/83/EC provide for a harmonized protection period regardless of the approval route utilized. The harmonized protection period is in total 10 years, including eight years of research data protection and two years of marketing protection. The effect is that the originator’s results can be the subject of a cross-referral application after eight years, but any resulting authorization cannot be exploited for a further two years. The rationale of this procedure is that the relevant particulars can, if the research data protection period has expired, be found on the originator’s file and used for assessment of the generic medicinal product. The 10-year protection period can be extended to 11 years where, in the first eight years post-authorization, the holder of the authorization obtains approval for a new indication assessed as offering a significant clinical benefit in comparison with existing products.
If the copy product does not meet the definition of a generic medicinal product or if bioequivalence could not be demonstrated through bioavailability studies or in case of certain types of changes in the active substance(s) or in the therapeutic indications, strength, pharmaceutical form or route of administration in relation to the reference medicinal product, Article 10(3) of Directive 2001/83/EC provides that the results of the appropriate pre-clinical studies or clinical trials must be provided by the applicant.
Well-established Medicinal Use
Under Article 10a of Directive 2001/83/EC, an applicant may, in substitution for the results of its own pre-clinical and clinical research, present detailed references to published literature demonstrating that the active substance(s) of a product have a well- established medicinal use within the community for at least ten years with recognized efficacy and an acceptable level of safety in terms of the conditions set out in Annex I of Directive 2001/83/EC. In that event, the test and trial results shall be replaced by appropriate scientific literature. The applicant is entitled to refer to a variety of different types of literature, including reports of clinical trials with the same active substance(s) and epidemiological studies that indicate that the constituent or constituents of the product have an acceptable safety/efficacy profile for a particular indication. However, use of the published literature exemption is restricted by stating that in no circumstances active substances be treated as having a well- established use if they have been used for less than 10 years from the first systematic and documented use of the substance as a medicinal product in the EU. Even after 10 years’ systematic use, the threshold for well-established medicinal use might not be met. European pharmaceutical law requires the competent authorities to consider among other factors the period over which a substance has been used, the amount of patient use of the substance, the degree of scientific interest in the use of the substance (as reflected in the scientific literature) and the coherence (consistency) of all the scientific assessments made in the literature. For this reason, different substances may reach the threshold for well-established use after different periods, but the minimum period is 10 years. If the applicant seeks approval of an entirely new therapeutic use compared with that to which the published literature refers, additional pre-clinical and/or clinical results would have to be provided.
| 94 |
Authorization Holder’s Consent
Under Article 10c of Directive 2001/83/EC, following the grant of a marketing authorization the holder of such authorization may consent to a competent authority utilizing the pharmaceutical, pre-clinical and clinical documentation that it submitted to obtain approval for a medicinal product to assess a subsequent application relating to a medicinal product possessing the same qualitative and quantitative composition with respect to the active substances and the same pharmaceutical form.
Law Relating to Pediatric Research
Regulation (EC) 1901/2006 (as amended by Regulation (EC) 1902/2006) was adopted on December 12, 2006. This Regulation governs the development of medicinal products for human use in order to meet the specific therapeutic needs of the pediatric population. It requires any application for marketing authorization made after July 26, 2008 in respect of a product not authorized in the European Community on January 26, 2007 (the time the Regulation entered into force), to include the results of all studies performed and details of all information collected in compliance with a pediatric investigation plan agreed by the Pediatric Committee of the EMA, unless the product is subject to an agreed waiver or deferral or unless the product is excluded from the scope of Regulation 1901/2006 (generics, hybrid medicinal products, biosimilars, homeopathic and traditional (herbal) medicinal products and medicinal products containing one or more active substances of well-established medicinal use) according to its Art. 9. Waivers can be granted in certain circumstances where pediatric studies are not required or desirable. Deferrals can be granted in certain circumstances where the initiation or completion of pediatric studies should be deferred until appropriate studies in adults have been performed. The EMA does not evaluate an application for market authorization that is not exempt from Regulation (EC) 1901/2006 if there is no agreed PIP, deferral or waiver. Moreover, this regulation imposes the same obligation from January 26, 2009 on an applicant seeking approval of a new indication, pharmaceutical form or route of administration for a product already authorized and still protected by a supplementary protection certificate granted under Regulation EC 469/2009 and its precursor Regulation (EEC) 1768/92 or by a patent that qualifies for the granting of such a supplementary protection certificate. The pediatric Regulation (EC) 1901/2006 also provides, subject to certain conditions, a reward for performing such pediatric studies, regardless of whether the pediatric results provided resulted in the grant of a pediatric indication. This reward comes in the form of an extension of six months to the supplementary protection certificate granted in respect of the product, unless the product is subject to orphan drug designation, in which case the 10-year market exclusivity period for such an orphan product is extended to 12 years. If any of the non-centralized procedures for marketing authorization have been used, the six-month extension of the supplementary protection certificate is only granted if the medicinal product is authorized in all member states.
Post-authorization Obligations
In the pre-authorization phase, the applicant must provide a detailed pharmacovigilance plan that it intends to implement post- authorization. An authorization to market a medicinal product in the EU carries with it an obligation to comply with many post- authorization organizational and behavioral regulations relating to the marketing and other activities of authorization holders. These include requirements relating to post-authorization efficacy studies, post-authorization safety studies, adverse event reporting and other pharmacovigilance requirements, advertising, packaging and labeling, patient package leaflets, distribution and wholesale dealing. The regulations frequently operate within a criminal law framework and failure to comply with the requirements may not only affect the authorization, but also can lead to financial and other sanctions levied on the company in question and responsible officers. EU pharmacovigilance legislation has been significantly modified by the Pharmacovigilance Directive, Dir. 2010/84/EU which amended the legal framework of pharmacovigilance for medicines marketed within the EU provided in Regulation (EC) No 726/2004 with respect to EU authorized medicinal products and in Directive 2001/83/EC with respect to nationally authorized medicinal products (including those authorized through the mutual recognition and decentralized systems). In addition, Commission Implementing Regulation (EU) No 520/2012 outlines the practical details to be respected by marketing authorization holders, national competent authorities and the EMA, and Commission Delegated Regulation (EU) No 357/2014 on post-authorization efficacy studies specifies the situations in which such studies may be required. Furthermore, EU good pharmacovigilance practice (GVP) rules apply. With these pharmacovigilance requirements, the financial and organizational burden on market authorization holders is significant, such as the obligation to maintain a pharmacovigilance system master file that applies to all holders of marketing authorizations granted in accordance with Directive 2001/83/EC or Regulation (EC) No 726/2004. Marketing authorization holders must furthermore collect data on adverse events associated with use of the authorized product outside the scope of the authorization. Pharmacovigilance for biological products and medicines with a new active substance is even stricter, as their authorization is subject to additional monitoring activities.
| 95 |
Any authorization granted by member state authorities, which within three years of its granting is not followed by the actual placing on the market of the authorized product in the authorizing member state, ceases to be valid (Art. 24 (4) and (5) Directive 2001/83/EC). When an authorized product previously placed on the market in the authorizing member state is no longer actually present on the market for a period of three consecutive years, the authorization for that product shall cease to be valid. The same two three-year periods apply to authorizations granted by the European Commission based on the centralized procedure (Art. 14 (4) and (5) Regulation (EC) 726/2004).
Other Countries
In addition to regulations in the United States, the EU, the UK and Israel, we are subject to a variety of other regulations governing clinical trials and commercial sales and distribution of drugs in other countries. Whether or not Aramchol or any other product candidate receive approval from the FDA, approval of such product candidates must be obtained by the comparable regulatory authorities of countries other than the United States before we can commence clinical trials or marketing of the product in those countries. The approval process varies from jurisdiction to jurisdiction, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials and product licensing vary greatly from country to country.
The requirements that we and our collaborators must satisfy to obtain regulatory approval by government agencies in other countries prior to commercialization of Aramchol or any other product candidate in such countries can be rigorous, costly and uncertain. In the European countries, UK, Canada and Australia, regulatory requirements and approval processes are similar in principle to those in the United States. Additionally, depending on the type of drug for which approval is sought, there are currently two potential tracks for marketing approval in the European countries: mutual recognition and the centralized procedure. These review mechanisms may ultimately lead to approval in all EU countries, but each method grants all participating countries some decision-making authority in product approval. The UK has a separate review period but for a transitional period until 31 December 2022, may rely on approvals under the EU mutual recognition and/or centralized procedure. Foreign governments also have stringent post-approval requirements including those relating to manufacture, labeling, reporting, record keeping and marketing. Failure to substantially comply with these on-going requirements could lead to government action against the product, us and/or our representatives.
Related Matters
From time to time, legislation is drafted, introduced and passed in governmental bodies that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA, MHRA or EMA and other applicable regulatory bodies to which we are subject. In addition, regulations and guidance are often revised or reinterpreted by the national agency in ways that may significantly affect our business and our therapeutic candidates. It is impossible to predict whether such legislative changes will be enacted, whether FDA, MHRA or EMA regulations, guidance or interpretations will change, or what the impact of such changes, if any, may be. We may need to adapt our business and therapeutic candidates and products to changes that occur in the future.
C. Organizational Structure
See “Item 4. Information on the Company—Historical Background and Corporate Structure” above.
| 96 |
D. Description of Property and Facilities
Our corporate headquarters are located at 16 Tiomkin Street, Tel Aviv, pursuant to a lease to occupy approximately 590 square meters of space. On March 22, 2015, GRD entered into the lease agreement with Mintz K. Construction Company for our corporate headquarters. We have since extended the option and extended the lease; most recently in March 2021. The lease expires on March 22, 2023, and we have an option to extend the lease for another year. The aggregate quarterly rental payment, together with adjustments and the maintenance fees, is approximately NIS 134,508 plus VAT (43,250 USD).
ITEM 4A. Unresolved Staff Comments.
None.
ITEM 5. Operating and Financial Review and Prospects.
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our financial statements and related notes that appear elsewhere in this annual report. In addition to historical financial information, the following discussion contains forward-looking statements that reflect our plans, estimates and beliefs. Our actual results could differ materially from those discussed in the forward-looking statements. Factors that could cause or contribute to these differences include those discussed below and elsewhere in this prospectus, particularly in the sections titled “Risk Factors” and “Cautionary Note Regarding Forward-Looking Statements.”
Overview
We are a clinical-stage biopharmaceutical company focused on the development of Aramchol, a liver targeted SCD1, modulator, first in class, novel, oral therapy for the treatment of NASH for variable populations. In September 2019, we initiated our Phase 3 pivotal ARMOR Study to evaluate the efficacy and safety of Aramchol in subjects with NASH and fibrosis. We are also collaborating with the Hebrew University in the development of Amilo-5MER, a 5 amino acid synthetic peptide.
To date, we have not generated revenue from the sale of any product, excluding the licensing revenue we recorded in connection with the Samil Agreement, and we do not expect to generate any significant revenue other than the amortization of the upfront payments under the license agreement with Samil and of the subsequent royalties and/or milestones that may be earned in connection with the Samil Agreement or potential other license Agreements, unless and until we commercialize Aramchol, or license the product to additional third parties. As of December 31, 2021, we had an accumulated deficit of approximately $168.2 million.
Our financing activities are described below under “Liquidity and Capital Resources.” Obtaining approval of an NDA, MMA, or other similar application is an extensive, lengthy, expensive and uncertain process, and the FDA, EMA, MHRA and other regulatory agencies may delay, limit or deny approval of Aramchol, Amilo-5MER or any other product candidate.
| 97 |
Financial Overview
To date, we have funded our operations primarily through proceeds from private placements and public offerings. At December 31, 2021, we had current assets of $36.1 million, which is mainly comprised of cash and cash equivalents of $2.9 million, restricted cash of $0.1 million, and short-term marketable securities of $31.9 million. This compares with current assets of $51.8 million at December 31, 2020, which is mainly comprised of cash and cash equivalents of $6.9 million, restricted cash of $0.1 million, short-term deposits of $3.8 million and short-term marketable securities of $40.1 million. We believe that such existing funds will be sufficient to continue our business and operations as currently conducted for more than 12 months from the date of issuance of this annual report. However, additional funding will be necessary to fund our ARMOR Study, our Amilo-5MER program and ongoing research and development work and to advance our product candidates through regulatory approval and into commercialization, if approved. We intend to obtain additional funding through debt or equity financings, governmental grants or through entering into collaborations, strategic alliances or license agreements to increase the funds available to support our operating and capital needs. Although we have been successful in raising capital in the past, there is no assurance that we will be successful in obtaining additional financing on terms acceptable to us. Specifically, the COVID-19 pandemic has significantly disrupted global financial markets, and may limit our ability to access capital, which could in the future negatively affect our liquidity. If funds are not available, we may be required to delay, reduce the scope of or eliminate research or development plans for, or commercialization efforts with respect to Aramchol, Amilo-5MER and/or our other pre-clinical and clinical programs. This may raise substantial doubts about our ability to continue as a going concern.
Revenues
We have entered into the Samil Agreement for the commercialization of Aramchol in Korea. Under the terms of the Samil Agreement, we have received upfront and milestone payments of $3.6 million, and may be eligible to receive up to approximately $4.5 million in additional payments for development and regulatory milestones for Aramchol in the licensed territories.
In accordance with ASC 606 the Company determined that the Agreement included a combined performance obligation representing the delivery of the exclusive license and completion of the ARREST study.
As of December 31, 2021, management evaluated the remaining clinical and regulatory milestones and determined that the variable consideration should not be recorded as revenue for the period ended December 31, 2021. The Company will re-evaluate the transaction price in each reporting period when events whose outcomes are resolved or other changes in circumstances occur that would indicate it is appropriate to recognize variable consideration as revenue
Costs and Operating Expenses
Our current costs and operating expenses consist of two components: (i) research and development expenses; and (ii) general and administrative expenses.
Research and Development Expenses
Our research and development expenses consist primarily of outsourced development expenses, salaries and related personnel expenses and fees paid to external service providers, patent-related legal fees, costs of pre-clinical studies and clinical trials and drug and laboratory supplies. We account for all research and development expenses as they are incurred. We expect our research and development expense to remain our primary expense in the near future as we continue to develop Aramchol and Amilo-5MER. Increases or decreases in research and development expenditures are primarily attributable to the number and/or duration of the pre-clinical and clinical studies that we conduct.
We expect that a substantial amount of our research and development expense in the future will be incurred in support of our current and anticipated pre-clinical and clinical development projects. Due to the inherently unpredictable nature of pre-clinical and clinical development studies, we are unable to estimate with any certainty the costs we will incur in the continued development of Aramchol, Amilo-5MER and any other potential product candidate. Clinical development timelines, the probability of success and development costs can differ materially from expectations. We currently expect to continue testing Aramchol and Amilo-5MER in pre-clinical studies for toxicology, safety and efficacy, and to conduct additional clinical trials for Aramchol and to initiate a first-in-human clinical study for Amilo-5MER.
| 98 |
While we are currently focused on advancing Aramchol’s and Amilo-5MER’s development, our future research and development expenses will depend largely on the duration of the ARMOR study, the number of enrolled patients, the clinical success of Aramchol, as well as ongoing assessments of the Aramchol’s commercial potential. As we obtain results from clinical trials, we may elect to discontinue or delay clinical trials for our product candidate in certain indications in order to focus our resources on more promising indications for such product candidate. Completion of clinical trials may take several years or more, but the length of time generally varies according to the type, complexity, novelty and intended use of a product candidate.
We expect our research and development expenses to increase in the future from current levels as we continue to advance our clinical product development into a pivotal stage trial and, potentially, the in-licensing of additional product candidates.
The lengthy process of completing clinical trials and seeking regulatory approval for Aramchol and Amilo-5MER or any other product candidate requires the expenditure of substantial resources. Any failure or delay in completing clinical trials, or in obtaining regulatory approvals, could cause a delay in generating product revenue and cause our research and development expenses to increase and, in turn, have a material adverse effect on our operations. Because of the factors set forth above, we are not able to estimate with any certainty when we would recognize any net cash inflows from our projects.
General and Administrative Expenses
General and administrative expenses consist primarily of compensation for employees in executive and operational roles, including finance/accounting, legal and other operating positions in connection with our activities. Our other significant general and administrative expenses include non-cash stock-based compensation costs and facilities costs (including the rental expense for our offices in Tel Aviv, Israel), professional fees for outside accounting and legal services, travel costs, investors relations, insurance premiums and depreciation. At this time, we do not anticipate that the effects of the COVID-19 pandemic will materially affect our general and administrative expense.
Financial Income, Net
Our financial income consists mainly of interest income from marketable debt securities and short-term deposits, as well as gains from realization of marketable debt securities and foreign currency gains. Our financial expense consists of fees associated with banking activities and losses from realization of marketable debt securities and foreign currency losses.
A. Results of Operations
The table below provides our results of operations for the year ended December 31, 2021 as compared to the year ended December 31, 2020.
| Year Ended December 31, | ||||||||
| 2020 | 2021 | |||||||
| (thousands) | ||||||||
| Research and development expenses | $ | 26,082 | $ | 27,220 | ||||
| General and administrative expenses | 4,128 | 5,661 | ||||||
| Operating loss | 30,210 | 32,881 | ||||||
| Financial income, net | (1,439 | ) | (414 | ) | ||||
| Net loss | $ | 28,771 | $ | 32,467 | ||||
| Comprehensive loss | $ | 28,534 | $ | 32,910 | ||||
| Basic and diluted net loss per share from continuing operations | $ | 1.35 | $ | 1.32 | ||||
| 99 |
Research and Development Expenses
Our research and development expenses amounted to approximately $27.2 million during the year ended December 31, 2021, representing an increase of approximately $1.1 million, or approximately 4%, compared to approximately $26.1 million for the year ended December 31, 2020. The increase primarily resulted from an increase in clinical studies in the amount of approximately $1.7 million partially offset by a decrease in drug development expenses in the amount of approximately $0.8 million.
General and Administrative Expenses
Our general and administrative expenses amounted to approximately $5.7 million for the year ended December 31, 2021, representing an increase of approximately $1.6 million, or 39%, compared to approximately $4.1 million for the year ended December 31, 2020. The increase primarily resulted from an increase in salaries and benefits of $0.8 million, as well as an increase in the cost of our D&O insurance policy premium of approximately $0.5 million.
Operating Loss
As a result of the foregoing research and development and general and administrative expenses, as well as our failure to generate substantial operating revenues, our operating loss for the year ended December 31, 2021 was approximately $32.9 million, representing an increase in our operating loss of approximately $2.7 million, or approximately 9%, compared to approximately $30.2 million for the year ended December 31, 2020.
Financial Income, Net
Our financial income, net, for the year ended December 31, 2021 was approximately $0.4 million, representing a decrease of approximately $1.0 million, or approximately 71%, compared to approximately $1.4 million for the year ended December 31, 2020. The decrease primarily resulted from a decrease in interest income from marketable debt securities and short-term deposits, as compared to such income for the comparable period in 2020.
Net Loss
Our net loss for the year ended December 31, 2021 was approximately $32.5 million, representing an increase of approximately $3.7 million, or approximately 13%, compared to approximately $28.8 million for the year ended December 31, 2020. The increase primarily resulted from the above-mentioned increase in research development expenses.
B. Liquidity and Capital Resources
Overview
To date, we have funded our operations primarily through proceeds from private placements and public offerings.
We have incurred substantial losses since our inception. As of December 31, 2021, we had an accumulated deficit of approximately $168.2 million and working capital (current assets less current liabilities) of approximately $30.2 million. Due to our expectation that we will continue to not generate substantial revenues for the foreseeable future, we expect that losses will continue for the foreseeable future.
| 100 |
As of December 31, 2021, we had cash and cash equivalents of approximately $2.9 million, restricted cash of $0.1 million, and marketable debt securities of approximately $31.9 million invested in accordance with our investment policy, totaling approximately $34.9 million, as compared to cash and cash equivalents of approximately $6.9 million, restricted cash of $0.1 million, short-term deposits of approximately $3.8 million and marketable debt securities of approximately $40.1 million, totaling approximately $51.0 million as of December 31, 2020. The decrease is mainly attributable to the $32.9 million negative cash flow from operating expenses during the year ended December 31, 2021, partially offset by the $17.4 million net raised from our ATM offering program and underwritten public offering.
The following table summarizes our significant contractual obligations at December 31, 2021.
| Less than 1 | ||||||||||||
| Total | year | 1 – 3 years | ||||||||||
| (in thousands) | ||||||||||||
| Facility leases (1) | $ | 376 | $ | 161 | $ | 215 | ||||||
| Car leases | 41 | 24 | 17 | |||||||||
| Total | $ | 417 | $ | 185 | $ | 232 | ||||||
We enter into contracts in the ordinary course of business with CROs for clinical trials and clinical supply manufacturing and with vendors for pre-clinical research studies and other services and products for operating purposes, which generally provide for termination within 30 to 90 days of notice, and therefore are cancelable contracts and not included in the Contractual Obligations table above. We have included as purchase obligations our commitments under agreements to the extent they are quantifiable and are not cancelable.
Other than as described above, we did not have any material commitments for capital expenditures, including any anticipated material acquisition of plant and equipment or interests in other companies, as of December 31, 2021.
Cash Flow from Operating Activities
We had negative cash flow from operating activities of approximately $32.9 million for the year ended December 31, 2021 as compared to a negative cash flow from operating activities of approximately $26.3 million for the year ended December 31, 2020. The negative cash flow from operating activities for the year ended December 31, 2021 was mainly attributable to our net loss of approximately $32.5 million.
Cash Flow from Investing Activities
We had positive cash flow from investing activities of approximately $11.4 million for the year ended December 31, 2021 as compared to a positive cash flow from investing activities of approximately $16.5 million for the year ended December 31, 2020. The positive cash flow from investing activities for the year ended December 31, 2021 was primarily due to the net maturity of short-term deposits in the amount of approximately $3.8 million, and the net sale of marketable debt securities in the amount of approximately $7.7 million.
| 101 |
Cash Flow from Financing Activities
We had positive cash flow from financing activities of approximately $17.4 million for the year ended December 31, 2021 as compared to a positive cash flow from financing activities of $0.8 million for the year ended December 31, 2020. The positive cash flow from financing activity for the year ended December 31, 2021 was due to proceeds from our ATM Offering and our public offering.
On May 15, 2020, we amended and restated the Sales Agreement dated December 22, 2017 between us and Stifel, Nicolaus & Company, Incorporated, or Stifel, to include Cantor Fitzgerald & Co., or Cantor as an additional sales agent for our “at the market offering” program, or the A&R Sales Agreement. During February 2021, we sold an additional 1,541,400 ordinary shares under the ATM offering program for total net proceeds of approximately $8.1 million. On March 25, 2021, we agreed with Cantor and Stifel to terminate, with immediate effect, the A&R Sales Agreement, among the Company, Stifel and Cantor.
On March 26, 2021, we entered into a new Sales Agreement with Cantor and Canaccord Genuity LLC as sales agents, or the Sale Agents, pursuant to which we may offer and sell our ordinary shares, par value NIS 0.01 per share, having an aggregate offering price of up to $50.0 million, from time to time through the Sales Agents. Under the new Sales Agreement, we may sell, from time to time, up to approximately $50.0 million of additional ordinary shares subject to limitations under the Baby Shelf Rule.
Current Outlook
Although we provide no assurance, we believe that our existing funds will be sufficient to continue our business and operations as currently conducted for more than 12 months from the date of issuance of this Annual Report on Form 20-F. However, additional funding will be necessary to fund our ARMOR Study, our Amilo-5MER program and ongoing research and development work and to advance our product candidates through regulatory approval and into commercialization, if approved. We intend to obtain additional funding through debt or equity financings, governmental grants or through entering into collaborations, strategic alliances or license agreements to increase the funds available to support our operating and capital needs. Although we have been successful in raising capital in the past, there is no assurance that we will be successful in obtaining additional financing on terms acceptable to us. Specifically, the COVID-19 pandemic has significantly disrupted global financial markets, and may limit our ability to access capital, which could in the future negatively affect our liquidity. If funds are not available, we may be required to delay, reduce the scope of or eliminate research or development plans for, or commercialization efforts with respect to Aramchol, Amilo-5MER and/or our other pre-clinical and clinical programs. This may raise substantial doubts about our ability to continue as a going concern.
The extent of our future capital requirements will depend on many other factors, including:
| ● | the progress and costs of our pre-clinical studies, clinical trials and other research and development activities; | |
| ● | the regulatory pathway of Aramchol, Amilo-5MER or any other product candidate; | |
| ● | the scope, prioritization and number of our clinical trials and other collaboration, research and development programs; | |
| ● | the amount of revenues and contributions we receive under future licensing, development and commercialization arrangements with respect to Aramchol, Amilo-5MER or any other product candidate; | |
| ● | the costs of the development and expansion of our operational infrastructure; |
| 102 |
| ● | the costs and timing of obtaining regulatory approval for Aramchol, Amilo-5MER or any other product candidate; | |
| ● | the ability of us, or our collaborators, to achieve development milestones, marketing approval and other events or developments under our potential future licensing agreements; | |
| ● | the costs of filing, prosecuting, enforcing and defending patent claims and other intellectual property rights; | |
| ● | the costs and timing of securing manufacturing arrangements for clinical or commercial production; | |
| ● | the costs of contracting with third parties to provide sales and marketing capabilities for us; | |
| ● | the costs of acquiring or undertaking development and commercialization efforts for any future products, product candidates or platforms; | |
| ● | the magnitude of our general and administrative expenses; | |
| ● | any cost that we may incur under future in- and out-licensing arrangements relating to Aramchol, Amilo-5MER or any other product candidate; | |
| ● | market conditions; and | |
| ● | the impact of the COVID-19 pandemic and the Russian invasion of Ukraine, which may exacerbate the magnitude of the factors discussed above. |
C. Research and Development, Patents and Licenses
For information concerning our research and development policies and a description of the amount spent during each of the last three fiscal years on company-sponsored research and development activities, see “Item 5. Operating and Financial Review and Prospects—Results of Operations.”
D. Trend Information
We are a development stage company and it is not possible for us to predict with any degree of accuracy the outcome of our research, development or commercialization efforts. As such, it is not possible for us to predict with any degree of accuracy any known trends, uncertainties, demands, commitments or events that are reasonably likely to have a material effect on our net sales or revenues, income from continuing operations, profitability, liquidity or capital resources, or that would cause reported financial information to not necessarily be indicative of future operating results or financial conditions. However, to the extent possible, certain trends, uncertainties, demands, commitments and events are in this “Operating and Financial Review and Prospects.”
E. Critical Accounting Policies and Estimates
We prepare our financial statements in accordance with U.S. GAAP. In doing so, we must make estimates and assumptions that affect our reported amounts of assets, liabilities and expenses, as well as related disclosure of contingent assets and liabilities. In some cases, we could reasonably have used different accounting policies and estimates. Changes in the accounting estimates are reasonably likely to occur from period to period. Accordingly, actual results could differ materially from our estimates. To the extent that there are material differences between these estimates and actual results, our financial condition or results of operations will be affected.
| 103 |
While our significant accounting policies are described in more detail in the notes to our audited consolidated financial statements appearing elsewhere in this Annual Report on Form 20-F we believe that the following accounting policies are those most critical to the judgments and estimates used in the preparation of our consolidated financial statements:
Accounting for stock-based compensation:
We grant equity-based awards under share-based compensation plans. We estimate the fair value of share-based payment awards using the Black-Scholes option valuation model. The Black-Scholes option valuation model requires the input of subjective assumptions, including price volatility of the underlying stock, risk-free interest rate, dividend yield, and expected life of the option. Share-based compensation expense is based on awards ultimately expected to vest, and therefore is reduced by expected forfeitures. Changes in assumptions used under the Black-Scholes option valuation model could materially affect our net loss and net loss per share.
Accounting for Marketable Debt Securities
Our debt securities are classified as available-for-sale and recorded at fair value. We determine the appropriate classification of investments in debt securities at the acquisition date and re-evaluates the classification at each balance sheet date. Unrealized gains and losses during the year, net of the related tax effect applicable to available-for-sale are excluded from income and reflected in other comprehensive income (loss) as a separate component of shareholders’ equity until realized. Prior to January 1, 2020, if a decline in fair value was deemed to be other-than-temporary, the investment was written down to its fair value and the amount of the write-down is recorded as an other-than-temporary impairment (“OTTI”) loss on the statement of operations. As the result of the adoption of Accounting Standards Update (“ASU”) 2016-13, Financial Instruments-Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instrument (“ASU 2016-13”) beginning on January 1, 2020, we instead assess the need at each period end to record an allowance for credit loss. Any portion of the market decline related to debt securities that is believed to arise from factors other than credit is recorded as a component of other comprehensive income (loss) rather than against income.
Revenue recognition
We have entered into the Samil Agreement for the commercialization of Aramchol in Korea. Under the terms of the Samil Agreement, we have received upfront and milestone payments, and may be eligible to receive additional payments for development and regulatory milestones. In accordance with ASC 606 we determined that the Agreement included a combined performance obligation representing the delivery of the exclusive license and completion of the ARREST study. We will re-evaluate the transaction price in each reporting period when events whose outcomes are resolved or other changes in circumstances occur that would indicate it is appropriate to recognize variable consideration as revenue
| 104 |
ITEM 6. Directors, Senior Management and Employees.
A. Directors and Senior Management.
Set forth below is information concerning the directors, senior management and executive officers of the Company as of April 15, 2022, the latest practicable date for inclusion in this annual report. The business address for each of our directors, senior management and corporate officers is c/o Galmed Pharmaceuticals Ltd., 16 Tiomkin St., Tel Aviv 6578317, Israel.
| Name | Age | Position | ||
| Allen Baharaff | 57 | President and Chief Executive Officer, Class II Director | ||
| Dr. Liat Hayardeny | 55 | Chief Scientist Officer | ||
| Doron Cohen | 55 | Chief Financial Officer | ||
| Yohai Stenzler | 39 | Chief Accounting Officer | ||
| Guy Nehemya | 37 | Chief Operating Officer and Data Protection Officer | ||
| David Sidransky, M.D.(1)(2)(3)(4)(5) | 61 | Lead Independent Director and Chairman of the R&D Committee, Chairman of our Nomination Committee, Chairman of our Remuneration Committee, and Class III director | ||
| Shmuel Nir(2)(3)(4)(5) | 60 | Class I Director | ||
| Amir Poshinski(2)(3)(4)(5) | 61 | Chairman of our Audit Committee, Class III director | ||
| Carol L. Brosgart, M.D. (1) (2) | 70 | Class I Director | ||
| Marshall Heinberg (2) | 65 | Class II Director |
| (1) | A member of our research & development committee. | |
| (2) | Independent director under applicable Nasdaq Capital Market and SEC rules, as affirmatively determined by our Board. | |
| (3) | A member of our audit committee. | |
| (4) | A member of our remuneration committee. | |
| (5) | A member of our nomination committee. |
Allen Baharaff, our President and Chief Executive Officer of our Board, co-founded the Group in 2000, served as the Chief Financial Officer of GHI from 2000 until January 2015, and has served as our Chief Executive Officer since January 2012 and as our President since March 2015. Previously, he held a senior executive position at Isramex Projects Ltd., an energy project financing company, and Managing Director of T+M Trusteeship & Management Services (Israel) Ltd., a subsidiary of a Swiss company providing trust and similar services. Since 2005, Mr. Baharaff serves as a Director of the Rubin Museum. Mr. Baharaff holds a Bachelor of Science degree in economics from the London School of Economics, University of London and LLB and MA degrees from Cambridge University. Since 1993, Mr. Baharaff has been a member of the Israel Bar Association.
| 105 |
Dr. Liat Hayardeny, our Chief Scientific Officer joined the Company in September 2016 bringing more than 17 years of experience in drug development at all stages as part of Teva Pharmaceuticals’ global Research and Development Division. Prior to joining Galmed, Dr. Hayardeny served as Teva’s Senior Director and Head of Research Scientific Affairs. In that capacity, Dr. Hayardeny established the scientific positioning of Teva’s innovative compounds. Additionally, Dr. Hayardeny was responsible for Teva’s relationship with institutions of higher education; managing Teva’s global research collaborations and publications. Dr. Hayardeny holds a Ph.D. from Sackler School of Medicine and an MBA from Recanati Business School at Tel Aviv University.
Doron Cohen, our Chief Financial Officer, joined the Company in February 2022, after providing the Company consulting services, including with respect to management of the Company’s funds, since 2018. Mr. Cohen brings more than twenty-five years of experience in the global financial markets, including significant experience on the buy side of life sciences companies. Since 2016, Mr. Cohen has served as Chief Executive Officer at Tangram Strategic Ltd., a boutique investment firm offering strategic solutions in economic & financial research, risk and regulation. Before that, between 1992 and 2017, Mr. Cohen served as Research Director at Gems Investments Research Ltd., an investment research company. During his tenure there, he provided research to hedge funds and managed three teams of analysts, focusing on investment research, operations research, and risk analysis. Prior to that from 1990 to 1992, Mr. Cohen served as Director of Public Relations at the World Union of Jewish Students. Mr. Cohen holds a Master of Science degree in management from Boston University, and a Bachelor’s degree in philosophy and political science from the University of New South Wales.
Yohai Stenzler, our Chief Accounting Officer, has served in such capacity since February 2022. Mr. Stenzler joined the Company in June 2014 as the Company’s corporate controller, and later on served as the Company’s Director of Finance. Between February 2017 to February 2022, Mr. Stenzler served as our Chief Financial Officer. Mr. Stenzler has six years of financial management experience as an accountant at the real estate department at Ernst & Young LLP, where he was involved in financing, taxes, auditing, advising and accounting of public and private companies, both domestic and international. Mr. Stenzler is a certified CPA and holds a MBA in Finance from Recanati Business School at Tel Aviv University, and a BA in Economics and Accounting from Ben-Gurion University of the Negev.
Guy Nehemya, our Chief Operating Officer and Data Protection Officer, has served in each role since January 2019 and March 2021, respectively. Between March 2017 and January 2019, Mr. Nehemya served as our Vice President, Operations. Mr. Nehemya joined the Company in October 2013 as the Company’s Director of Operations, after completing his internship at Agmon, Rosenberg, HaCohen & Co. Law Offices. Mr. Nehemya was a key member of management during the Company’s initial public offering and execution thereof. Mr. Nehemya holds a LL.B. from the College of Management and is currently completing his MBA degree at the IDC Herzliya. Mr. Nehemya has been a member of the Israeli Bar Association since 2012.
David Sidransky, M.D., the chairman of our Nomination, Remuneration and R&D Committees, joined our Board in June 2014, originally as an external director. Dr. Sidransky is a renowned oncologist and research scientist named and profiled by TIME magazine in 2001 as one of the top physicians and scientists in America, recognized for his work with early detection of cancer. He serves as the Director of the Head and Neck Cancer Research Program at the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University. He is a Professor of Oncology, Otolaryngology, Cellular & Molecular Medicine, Urology, Genetics, and Pathology at Johns Hopkins University and Hospital. Dr. Sidransky has written over 600 peer-reviewed publications and has contributed to more than 60 cancer reviews and chapters. Dr. Sidransky is a founder of a number of biotechnology companies and holds numerous biotechnology patents. He has been the recipient of many awards and honors, including the 1997 Sarstedt International prize from the German Society of Clinical Chemistry, 1998 Alton Ochsner Award Relating Smoking and Health by the American College of Chest Physicians and the 2004 Hinda Rosenthal Award and 2017 Team Award presented by the American Association of Cancer Research. Dr. Sidransky has served as Vice Chairman of the Board of Directors of ImClone. He is Chairman of the Board of Advaxis Inc. (Nasdaq: ADXS), and is a lead director at Champions Oncology and on the board of directors of Orgenesis (Nasdaq: ORGS), Ayala Pharma (Nasdaq: AYLA) andAscentage Pharma. He is serving and has served on scientific advisory boards of corporations and institutions, including Amgen, MedImmune, Roche and Veridex, LLC (a Johnson & Johnson diagnostic company), among others. In addition, Dr. Sidransky served as Director of American Association for Cancer Research from 2005 to 2008. Dr. Sidransky received his B.A. from Brandeis University and his M.D. from the Baylor College of Medicine.
| 106 |
Shmuel Nir, a director of the Company since 2007, serves as President and Chief Executive Officer of Tushia Consulting Engineers Ltd., an investment and management services company. From January 2001 to January 2016, Mr. Nir served as Chairman of the board of directors of Matan Digital Printers Ltd. From March 1998 to January 2008, he served as President and Chief Executive Officer of Macpell Industries Ltd., a leading industrial group. Between January 1991 and March 1998, Mr. Nir was an Executive Vice President of Operations at Macpell Industries Ltd. and President and Chief Executive Officer of two of its subsidiaries, New Net Industries Ltd. and New Net Assets Ltd. Prior to January 1991, Mr. Nir had held various positions with Intel Corporation in Jerusalem, Israel and Tefen Management Consulting. Between 1999 and 2006, Mr. Nir served as managing partner at Spring Venture Capital Fund. Mr. Nir holds a B.Sc. in Industrial Engineering and Management from the Technion - Israel Institute of Technology in Haifa, which was awarded in 1989.
Amir Poshinski, joined our Board in June 2020. Mr. Poshinski is an entrepreneur with over 20 years’ management and leadership experience across multiple industries, including technology, biotechnology, banking and real estate. Mr. Poshinski is the owner of DAP Holdings, through which he has acted since 2010 as a management consultant and strategic advisor to global companies. Mr. Poshinski currently serves as a member of the advisory board of Benson Oak Ventures, a venture focused fund, as well as several other private companies. Prior to 2010, Mr. Poshinski served as Deputy CEO of Primsa Investment House, which at the time was Israel’s largest investment house, Deputy CEO of Discount Mortgage Bank, the real estate lending arm of one of Israel’s largest banks, VP of Marketing at Comverse, a telecommunications software company that was listed on Nasdaq, VP Marketing, Sales and Advertising of Mifal Hapayis, Israel’s national lottery, and VP and Deputy CEO of the Economic Company of the Israeli Local Authorities Association. Mr. Poshinski previously served on the board of directors of each of TAS-AGT (a TATA joint venture), Excellence Nessuah Mutual Funds, and Therapix Biosciences (Nasdaq: TRPX) as well as several other private companies. Mr. Poshinski holds a B.A. in Business Administration and Marketing from the New York Institute of Technology.
Carol L. Brosgart, M.D. joined our Board on June 7, 2017. Dr. Brosgart served as a member of Tobira Therapeutics’s Board of Directors from 2009 until it was acquired by Allergan in 2016 and on the Board of Juvaris, a vaccine company acquired by Bayer. Since January 2018, she serves on the Board of Directors of Abivax, a biotechnology company, headquartered in Paris, working on HIV Cure and inflammatory diseases. Since June 2021, Dr. Brosgart also serves on the Board of Intrivo Diagnostic and Mirum Pharmaceuticals (Nasdaq: MIRM) since June 2021. Dr. Brosgart serves as a consultant to Dynavax, Allergan and a number of biotechnology companies in the areas of liver diseases and infectious diseases and on the Board of Enochian, focusing on HIV Cure. Dr. Brosgart currently serves on the Steering Committee of the National Viral Hepatitis Roundtable, the Executive Committee of the Forum for Collaborative Research, the Steering Committee of the HBV Cure Group at the Forum, and is on the Board of Directors of the Hepatitis B Foundation and the Northern California American Liver Foundation and the Board of Berkeley Community Scholars. She is active in the public policy arena for AASLD and IDSA/HIVMA. Dr. Brosgart served as Senior Advisor on Science and Policy to the Division of Viral Hepatitis at the CDC and to the Viral Hepatitis Action Coalition at the CDC Foundation from 2011 to 2013. Dr. Brosgart has also served as a member of the clinical faculty of the School of Medicine at the University of California, San Francisco for the past four decades, where she is a Clinical Professor of Medicine, Biostatistics and Epidemiology in the Division of Global Health and Infectious Diseases. In 2011, Dr. Brosgart served as Chief Medical Officer at biotechnology company Alios BioPharma, Inc. Prior to Alios, Dr. Brosgart served as Senior Vice President and Chief Medical Officer of Children’s Hospital & Research Center in Oakland, California, from 2009 until February 2011. Previously, she served for eleven years, from 1998 until 2009, at the biopharmaceutical company Gilead Sciences, Inc., where she held a number of senior management roles, most recently as Vice President, Public Health and Policy and earlier as Vice President, Clinical Research and Vice President, Medical Affairs. Prior to Gilead, Dr. Brosgart was the Medical Director of the East Bay AIDS Center in Berkeley, California (1987-1998) and the Medical Director of the Central Health Center for the Alameda County Public Health Department (1978-1987). Dr. Brosgart received a B.S. in Community Medicine from the University of California, Berkeley and received an M.D. from the University of California, San Francisco. Her residency training was in pediatrics, public health and preventive medicine at UCSF and UC Berkeley School of Public Health. She has published extensively in the areas of HIV, HBV, CMV, and liver disease.
| 107 |
Marshall Heinberg joined our Board on October 14, 2018. Mr. Heinberg has extensive experience relevant to us and insight into the global capital markets and has worked with several life science and technology companies. From 2020 to 2022, Mr. Heinberg served as a Chairman of the Board of PAE Inc, which was acquired by Amentum Government Services Holdings LLC. Mr. Heinberg is the founder and Managing Director of MAH Associates, LLC, which provides strategic advisory and consulting services to various companies, including for the Company from 2013 until September 2018. Mr. Heinberg also serves on the Board of Union Carbide Corporation (subsidiary of Dow Chemical) and of ChannelAdvisor (NYSE: ECOM), since July 2019 and December 2019, respectively and appointed as Chairman of Costume Truck Once Source in April 2020. From April 2017 to December 2019, Mr. Heinberg served on the board of directors of Ecology and Environment (Nasdaq: EEI) and was its Executive Chairman of the Board of Directors until the time that EEI was acquired by WSP. Between January 2010 and March 2021, Mr. Heinberg served on the board of directors of Universal Biosensors (UBI.AX). Mr. Heinberg was a Senior Advisor to Burford Capital (NYSE;BUR) until July 2020. Mr. Heinberg began his investment banking career in 1987 in the Corporate Finance Division of Oppenheimer & Co, Inc., which was acquired by Canadian Imperial Bank of Commerce (CIBC) in 1997. Mr. Heinberg served as Head of the Investment Banking Department and as a Senior Managing Director of Oppenheimer & Co. Inc. from 2008 until 2012, and as the U.S. Head of Investment Banking at CIBC World Markets from 2001 until 2008. Prior to joining Oppenheimer, Mr. Heinberg practiced corporate law for approximately four years. Mr. Heinberg has a B.S. in economics from the Wharton School at the University of Pennsylvania and a J.D. from Fordham Law School.
There are no family relationships between any director or executive officer. There are no arrangements or understandings with major shareholders, customers, suppliers or others, pursuant to which any director or executive officer was selected as a director or member of senior management, as the case may be.
Scientific Advisory Board
We seek advice from our Scientific Advisory Board generally on scientific and medical matters. Our Scientific Advisory Board includes the following: Professor Vlad Ratziu from the University Pierre et Marie Curie in Paris, France and coordinator of the EU FP7 FLIP consortium; Professor Scott Friedman from the Icahn School of Medicine at Mount Sinai in New York, United States; Professor Arun Sanyal, from the Virginia Commonwealth University in Richmond, Virginia; Professor Jose Mato, from CIC bioGUNE Spain; Professor Shomron Ben-Horin, Chief of the Gastroenterology Department at Sheba Medical Center and Prof. Stephen B. Hanauer from Northwestern Feinberg School of Medicine.
B. Compensation.
Certain Approvals Required for Office Holders’ Compensation of the Companies Law
Pursuant to the Companies Law, the Company is required to adopt a compensation policy regarding the terms of office and employment of its Office Holders (as such terms are defined below), which includes exemption and release of the Office Holders from liability for breach of his or her duty of care to the Company, an undertaking to indemnify the Office Holder, post factum indemnification or insurance; any grant, payment, remuneration, compensation, or other benefit provided in connection with termination of service; and any benefit, other payment or undertaking to provide any payment as aforesaid, or the Terms of Office and Employment. The Company’s current compensation policy with respect to the Terms of Office and Employment of the Company’s Office Holders, or the Compensation Policy, was last approved by the Board in June 22, 2020 after considering the recommendations of the remuneration committee and was adopted by the Company’s shareholders in August 2020.
The term ‘Office Holder’ as defined in the Companies Law includes a general manager, chief business manager, deputy general manager, vice general manager, any other person fulfilling or assuming the responsibilities of any of the foregoing positions without regard to such person’s title, as well as a director, or a manager directly subordinate to the general manager or the chief executive officer.
| 108 |
Pursuant to the Companies Law, arrangements between the Company and its Office Holders must generally be approved by the remuneration committee and the Board and be consistent with the Compensation Policy. However, under certain circumstances, the Company may approve an arrangement that is not consistent with the Compensation Policy, if such arrangement is approved by a majority of the Company’s shareholders, provided that (i) such majority includes a majority of the votes cast by shareholders who are not controlling shareholders and who do not have a personal interest in the matter, present and voting (abstentions are disregarded), or (ii) the votes cast by shareholders who are not controlling shareholders and who do not have a personal interest in the matter who were present and voted against the arrangement constitute two percent or less of the voting power of the company, or the Special Majority.
The terms of office and employment of directors (including an officer who is a director but is not a controlling shareholder) further require the approval of the shareholders by a simple majority in addition to the approval of the Compensation Committee and the Board, in that order, and under certain circumstances, a Special Majority; with respect to a chief executive officer or an officer who is a controlling shareholder, the approval of the shareholders must be made by the Special Majority. In addition, under certain circumstances, a company may be exempt from receiving the shareholders’ approval with respect to the Terms of Office and Employment of a non-affiliated candidate for chief executive officer.
Under certain circumstances, if the terms of office and employment of Office Holders (who are not directors or controlling shareholders) are not approved by the shareholders, where such approval is required, the remuneration committee and the Board may subsequently override the resolution of the shareholders following a new discussion of the matter and for specified reasons. In addition, amendment of terms of office and employment of Office Holders (who are not directors or controlling shareholders) requires the approval of the remuneration committee only, if the remuneration committee determines that the amendment is not material.
Aggregate Executive Compensation
The aggregate compensation, including share-based compensation, paid by us to all of our Office Holders as a group, with respect to the year ended December 31, 2021, was approximately $4.1 million. This amount includes approximately $0.4 million set aside or accrued to provide pension, severance, retirement, vacation or similar benefits or expenses, but does not include business travel, relocation, professional and business association dues and expenses reimbursed to Office Holders, and other benefits commonly reimbursed or paid by companies in our industry. In addition to the six current members of the Board (including the Company’s President and Chief Executive Officer), the Company considers seven other individuals, namely three former directors, the former Chief Medical Officer, Chief Scientist Officer, Chief Financial Officer and the Chief Operating Officer, to have been Office Holders in 2021.
As of December 31, 2021, options to purchase 2,152,867 of our ordinary shares granted to our Office Holders as a group were outstanding, of which options to purchase 1,549,117 of our ordinary shares have vested, with a weighted average exercise price of $5.77 per ordinary share.
For outstanding equity-based awards granted to our Office Holders, see below under “Item 6. Directors, Senior Management and Employees—E. Share Ownership—Certain Information Concerning Equity Awards to Office Holders.”
| 109 |
Individual Compensation of Covered Executives
The following table sets forth the compensation granted to the four most highly compensated Office Holders during or with respect to the year ended December 31, 2021. All amounts reported in the table reflect the cost to the Company, as recognized in its financial statements for the year ended December 31, 2021. The four individuals for whom disclosure is provided are referred to herein as “Covered Executives.”
| Information Regarding the Covered Executives | Compensation for Services(1) | |||||||||||||||||||||||
| Benefits | ||||||||||||||||||||||||
| Base | and | Cash | Equity-Based | |||||||||||||||||||||
| Salary | Perquisites | Bonus | Compensation | Other | ||||||||||||||||||||
| Name and Principal Position(1) | ($) | ($)(2) | ($)(3) | ($)(4) | ($)(5) | Total ($) | ||||||||||||||||||
| Allen Baharaff (President and Chief Executive Officer) | 631,579 | 204,511 | 834,964 | 803,527 | 40,000 | 2,514,580 | ||||||||||||||||||
| Dr. Liat Hayardeny (Chief Scientific Officer) | 205,077 | 57,938 | - | 159,107 | - | 421,583 | ||||||||||||||||||
| Yohai Stenzler (Chief Accounting Officer) | 129,288 | 43,823 | - | 156,684 | - | 329,795 | ||||||||||||||||||
| Guy Nehemya (Chief Operating Officer and Data Protection Officer) | 129,288 | 39,640 | - | 156,677 | - | 325,604 | ||||||||||||||||||
| (1) | The above-mentioned executives are all full-time employee of the Company. Cash compensation amounts denominated in currencies other than the Dollar were converted into Dollars at an exchange rate of NIS 3.23 = $1.00, which reflects the average conversion rate for fiscal year ended December 31, 2021. | |
| (2) | Amounts reported in this column include benefits and perquisites, including those mandated by applicable law. Such benefits and perquisites may include, to the extent applicable to the Covered Executives, payments, contributions and/or allocations for savings funds, pension, severance, vacation, car allowance, risk insurance (e.g., life, disability, accident), telephone, convalescence pay, payments for social security and other benefits and perquisites consistent with the Company’s policies. | |
| (3) | Amounts reported in this column refer to the cash bonuses provided by the Company with respect to 2021, which have been provided for in the Company’s financial statements for the year ended December 31, 2021 (including if such bonuses were paid in 2022). They exclude bonuses paid in 2021 which were provided for in the Company’s financial statements for previous years. Cash bonuses are paid in accordance with the Company’s 2021 Annual Cash Bonus Plan and are intended to promote the Company’s work plan and business strategy by rewarding officers for achievement of the Company’s business and financial goals through teamwork and collaboration. Key performance indicators which are factored into cash bonus determinations are based both on personal evaluation and as well, individual specific and may include: (i) major progress in research and development stages, (ii) the execution of in/out-license transactions, (iii) the execution of strategic collaboration agreements, and (iv) raising funds throughout public offering or a private placement. |
| 110 |
| (4) | Amounts reported in this column represent the expense recorded in the Company’s financial statements for the year ended December 31, 2021 with respect to equity-based compensation. Assumptions and key variables used in the calculation of such amounts are discussed in Note 10 to the Financial Statements. For outstanding equity-based awards granted to Covered Executives see below under “Item 6. Directors, Senior Management and Employees—E. Share Ownership—Certain Information Concerning Equity Awards to Office Holders.” | |
| (5) | Amounts reported in this column include payments made with respect to the year 2021 and recorded in the financial statements for the year ended December 31, 2021 relating to directors’ fees. |
Compensation of Directors
As approved by our shareholders at our 2020 annual meeting of shareholders, in connection with their services as directors of the Company, each of our directors from time to time, is entitled to an annual payment of $40,000, plus value-added tax, or VAT, if applicable, and with respect to an expert external director (if applicable), $50,000 plus VAT, payable quarterly at the end of each quarter, and, upon first becoming a member of the Board. As approved by our shareholders at our 2021 annual meeting of shareholders, each of our non-management directors will receive a grant of options to purchase 20,000 ordinary shares, or the Director Options. The Director Options would be granted under the 2013 Plan, at an exercise price equal to the average price of our ordinary shares on the Nasdaq market in the 30 trading days prior to the appointment by the Board or election by the shareholders (as applicable), and would vest over a period of three years, such that the Director Options will vest with respect to 1/3 of the underlying Ordinary Shares on the first anniversary of the grant (i.e., the date of appointment by the Board or election by the shareholders, as applicable), and thereafter, the Director Options will vest with respect to the additional 2/3 of the underlying ordinary shares on an equally quarterly basis, provided that each non-management director continues to serve as a director of us or our affiliates throughout each such vesting date. All unvested options held by a non-management director in office will automatically vest and become exercisable upon the consummation of a Transaction, as such term is defined in the 2013 Plan. The grant is subject to the execution by each director of an option agreement with us confirming the terms and conditions applying to the grant.
Our Board has determined that each of Mr. Nir, Mr. Poshinski, Mr. Heinberg and Dr. Sidransky are entitled to receive compensation as an ‘expert external director’. The compensation of external directors is also subject to the provisions of the Israeli regulations promulgated pursuant to the Companies Law governing the terms of compensation payable to external directors. See also “Item 6. Directors, Senior Management and Employees—C. Board Practices—External Directors” and “Item 7. Major Shareholders and Related Party Transactions—C. Related Party Transactions” below.
For the outstanding equity-based awards granted to our directors, see below under “Item 6. Directors, Senior Management and Employees—E. Share Ownership—Certain Information Concerning Equity Awards to Office Holders.”
Employment Agreements and Arrangements with Directors and Related Parties
We entered into written employment agreements with each of our executive officers. These agreements provide for notice periods of varying duration for termination of the agreement by us or by the relevant executive officer, during which time the executive officer will continue to receive base salary and benefits. These agreements also contain customary provisions regarding non-competition, confidentiality of information and assignment of inventions. However, the enforceability of the non-competition and assignment of inventions provisions may be limited under applicable law. See “Item 3. Key Information—Risk Factors—Risks Related to Our Business, Industry and Regulatory Requirements.”
| 111 |
Employment Agreement with Our President and Chief Executive Officer
On December 30, 2013, we entered into a personal employment agreement with our controlling shareholder, Mr. Allen Baharaff who serves as our president and chief executive officer and as the chairman of our Board, as amended on March 15, 2016, July 20, 2017, August 1, 2019, and August 30, 2021 which provides that Mr. Baharaff’s terms of office and employment are for an undefined term, subject to re-approval under the Companies Law and termination in accordance with the terms of the employment agreement.
Under the current terms of his employment agreement, Mr. Baharaff is entitled to a gross monthly salary of NIS 170,000, following shareholder approval which was obtained at our 2021 annual meeting of shareholders. In addition, Mr. Baharaff will be entitled to an annual cash bonus based on achievement of qualitative and quantitative performance goals and objectives. As approved at our 2021 annual meeting of shareholders the annual cash bonus amount can be of up to six times Mr. Baharaff’s monthly base salary, and the actual bonus paid in a given year shall be determined based on the achievement of certain qualitative and quantitative performance goals and objectives set by our remuneration committee and Board, which effective as of January 1, 2022, does not require further shareholder approval. Mr. Baharaff’s performance goals and objectives for the year 2021 were approved at our 2021 annual meeting of shareholders.
Previously, Mr. Baharaff was entitled to receive the following bonuses (i) upon execution of a Strategic Agreement (as defined below), and subject to the discretion of the Board, a cash bonus in an amount of up to twelve times his monthly base salary. A “Strategic Agreement” means: a license agreement or any other strategic agreement (i.e. research and development, manufacture, distribution, etc.) for the U.S., Europe, Japan or China; (ii) upon consummation of a fund raising (excluding funds received from a Strategic Agreement), and, subject to the discretion of the Board, a cash bonus in an amount of up to ten times his monthly base salary if the funds received by the Company are between $8 Million to $10 million and up to twelve times his monthly base salary if the funds received by the Company are $10 million or more; (iv) upon a Change of Control Event (as defined below), and, subject to the discretion of the Board, a cash bonus in an amount of up to twelve times his monthly base salary, a “Change of Control Event” means: (a) the acquisition of the Company by another entity or individual or group of individuals by means of any transaction or series of related transactions (including, without limitation, any reorganization, merger, share purchase or consolidation), unless the Company’s shareholders of record as constituted immediately prior to any such transaction will, immediately after such transaction (by virtue of securities issued as consideration for the Company’s share capital, assets or otherwise) hold more than 50% of the voting power of the surviving or acquiring entity; or (b) a sale of all or substantially all of the assets of the Company.
Mr. Baharaff will also be entitled to the following equity based compensation: (i) in the event that our options are cashed-out upon a Change of Control Event, all unvested options granted to Mr. Baharaff will vest immediately prior to the consummation of the Change of Control Event; (ii) if upon a Change of Control Event (a) Mr. Baharaff’s employment as chief executive officer of the Company or the surviving entity is terminated within twelve months as of the Change of Control Event, and (b) unvested options are replaced for new options of the surviving entity as part of the Change of Control Event with a vesting schedule and terms identical to the replaced options, or the Replacement Options, then (x) all unvested Replacement Options granted to Mr. Baharaff will vest immediately prior to the termination of Mr. Baharaff’s employment, and (y) Mr. Baharaff’s Replacement Options will be exercisable until the earlier of (a) two years from termination, and (b) expiration of the Replacement Options.
Mr. Baharaff will also receive other benefits required under Israeli law or that are customary for senior executives in Israel such as confidentiality, reimbursement of expenses, payment for absence days, sick leave, pension and/or a manager’s insurance policy and study fund.
Mr. Baharaff is also entitled to accumulate vacation days for no more than two years. As approved at our 2021 annual meeting of shareholders, unused accumulated vacation days that exceed the number of vacation days that may be accumulated over a two-year period (currently, 48 vacation days) shall be redeemed once a year, on March 1, provided that the redemption will not result in the number of accumulated vacation days following the redemption being less than 48 days, or as otherwise required by law. Accumulated vacation days shall also be redeemed in the event of termination of employment of Mr. Baharaff.
| 112 |
Mr. Baharaff’s employment agreement is terminable by either party upon six months prior written notice, or Prior Notice Period, and contains customary provisions regarding noncompetition, confidentiality of information and assignment of inventions. Upon termination, provided such termination was not for cause, Mr. Baharaff shall be entitled, in addition to the Prior Notice Period, to a payment in an amount of up to twelve times his monthly base salary, to be paid in twelve equal monthly installments, in exchange for Mr. Baharaff’s undertaking not to compete with the Company for a period of twelve months, or Non-Compete Grant. Other than in case of resignation by Mr. Baharaff, excluding resignation for a Good Reason Event (as defined below), or termination for cause: (i) all Mr. Baharaff’s unvested options will vest upon termination; and (ii) unexercised options granted to Mr. Baharaff may be exercised until the earlier of (a) two years from his termination, and (b) expiration of his options. A “Good Reason Event” means: any of the following events, provided that the event is effected by the Company without the written consent of Mr. Baharaff: (i) a material reduction or adverse change in Mr. Baharaff’s authority, duties or responsibilities; (ii) a reduction in Mr. Baharaff’s monthly base salary, other than a reduction of no more than 10% of his then current monthly base salary as part of an across the board reduction in all salaries for employees of the Company; (iii) a material breach by the Company of Mr. Baharaff’s employment agreement or any other agreements pertaining directly to Mr. Baharaff’s compensation or employment or (iv) death, disability or severe illness. Upon termination for cause by the Company, Mr. Baharaff shall not be entitled to any Prior Notice Period, Non-Compete Grant or any other payment, and any unvested outstanding equity awards shall terminate immediately upon the date of such termination for cause.
For cash bonuses granted to Mr. Baharaff see “Item 6. Directors, Senior Management and Employees— B. Compensation—Individual Compensation of Covered Executives.” For outstanding equity-based awards granted to Mr. Baharaff see below under “Item 6. Directors, Senior Management and Employees—E. Share Ownership—Certain Information Concerning Equity Awards to Office Holders.”
C. Board Practices.
We are incorporated in Israel, and, therefore, we are subject to various corporate governance practices under Israeli law relating to such matters as external directors (if required), independent directors, audit committees, remuneration committees and internal auditors. These Israeli law requirements are in addition to the requirements of the Nasdaq Listing Rules and other relevant provisions of U.S. securities laws. Under such Nasdaq Listing Rules, a foreign private issuer may generally follow its home country practices for corporate governance in lieu of such comparable listing rules’ requirements, except for certain matters such as composition and responsibilities of the audit committee and the SEC-mandated standards for the independence of its members. See below under “Item 16G. Corporate Governance” for further information.
Membership of the Board
Our Articles provide that the minimum number of members of the Board is three and the maximum number of members is eleven. The Board is presently comprised of six members. Under the Regulation, companies with no controlling shareholder whose shares are listed for trading on specified exchanges outside of Israel, including the Nasdaq Capital Market, may adopt exemptions from various corporate governance requirements of the Companies Law so long as the company satisfies the applicable foreign country laws and regulations, including applicable stock exchange rules, that apply to companies organized in that country relating to the appointment of independent directors and the composition of audit and compensation committees. Such exemptions include an exemption from the requirement to appoint external directors and the requirement that an external director be a member of certain committees.
| 113 |
In March 2020, our Board adopted the exemption under the Regulation, and our directors then in office who were elected and classified as external director, Tali Yaron-Eldar and Dr. David Sidransky, were no longer classified as such under the Companies Law. The transition rules set forth under the Regulation provide that such former external directors have the right to remain in office as company directors at their option after the exemption under the Regulation is adopted until the earlier of such director’s original end of term of office or the second annual meeting of shareholders after the adoption of the exemption under the Regulation. Ms. Yaron-Eldar’s and Dr. Sidransky’s term of office expired on June 12, 2020. On May 12, 2020, our Board extended Dr. Sidransky’s term as director as of June 12, 2020 through the end of the annual meeting of our shareholders which was held on August 13, 2020, and at our annual meeting of our shareholders, Dr. Sidransky’s term was further extended until the annual general meeting to be held in 2023, as a Class III director. Ms. Yaron-Eldar’s term was not extended.
On June 16, 2020, our Board appointed Mr. Amir Poshinski as a Class III director, and at our 2020 annual meeting of our shareholders, Mr. Poshinski was elected to serve as a Class III director until the annual general meeting to be held in 2023.
On July 15, 2021, our Board recommended to re-elect each of Prof. Carol Brosgart and Mr. Shmuel Nir as Class I directors, and at our 2021 annual meeting of our shareholders, Prof. Carol Brosgart and Mr. Shmuel Nir’s term was extended until the close of the annual general meeting to be held in 2024, as Class I directors.
The minimum and maximum number of directors may be changed, at any time and from time to time, by a majority vote of our directors then in office, provided that no decrease in the number of directors shall shorten the term of any incumbent director. Under our Articles, the Board consists of three classes of directors which are appointed for fixed terms of office in accordance with the Companies Law and our Articles, with one class being elected each year for a term of approximately three years by our shareholders at our annual general meeting.
Directors so elected cannot be removed from office by the shareholders until the expiration of their term of office. The directors do not receive any benefits upon the expiration of their term of office.
The three classes of directors are Class I Directors, Class II Directors and Class III Directors. Shmuel Nir and Dr. Carol Brosgart serve as our Class I Directors until the close of the annual general meeting to be held in 2024; Allen Baharaff and Marshall Heinberg serves as our Class II Directors until the close of the annual general meeting to be held in 2022; and Dr. David Sidransky and Amir Poshinski serve as our Class III Directors until the close of the annual general meeting to be held in 2023.
In accordance with the Articles, any vacancies on the Board of, including unfilled positions, may be filled by a vote of a majority of the directors then in office, and each director chosen in this manner would hold office until the next annual general meeting of the Company (or until the earlier termination of his or her appointment as provided for in the Companies Law or the Articles).
Any amendment of our Articles regarding the election of directors, as described above, require the affirmative vote of at least 75% of the voting rights in the Company, represented personally or by proxy and voting thereon at a general meeting. See “Item 6. Directors, Senior Management and Employees—C. Board Practices—External Directors” for a description of the procedure for the election of external directors.
A nominee for service as a director in a public company may not be elected without submitting a declaration to the company, prior to election, specifying that he or she has the requisite qualifications to serve as a director, independent director or external director (if required), as applicable, and the ability to devote the appropriate time to performing his or her duties as such.
A director, who ceases to meet the statutory requirements to serve as a director, external director or independent director, as applicable, must notify the company to that effect immediately and his or her service as a director will expire upon submission of such notice.
| 114 |
Alternate Directors
Our Articles provide, as allowed by the Companies Law, that any director may, subject to the conditions set thereto, appoint a person as an alternate to act in his place, to remove the alternate and appoint another in his place and to appoint an alternate in place of an alternate whose office is vacated for any reason whatsoever. Under the Companies Law, a person who is not qualified to be appointed as a director, a person who is already serving as a director or a person who is already serving as an alternate director for another director, may not be appointed as an alternate director. Nevertheless, a director who is already serving as a director may be appointed as an alternate director for a member of a committee of the board of directors so long as he or she is not already serving as a member of such committee. A person who is not qualified to be appointed as an independent director, pursuant to the Companies Law, may not be appointed as an alternate director of an independent director qualified as such under the Companies Law. Unless the appointing director limits the time or scope of the appointment, the appointment is effective for all purposes until the appointing director ceases to be a director or terminates the appointment.
External Directors
Generally, unless a regulatory relief is available, under the Companies Law and the regulations promulgated pursuant thereto, Israeli companies whose shares have been offered to the public, or that are publicly traded outside of Israel, which we refer to as a public company, are required to appoint at least two natural persons as “external directors.”
No person may be appointed as an external director if such person is a relative of a controlling shareholder or if such person, a relative, partner or employer of such person, or anyone to whom such person is directly or indirectly subordinate, or any entity under such person’s control, has or had, on or within the two years preceding the date of such person’s appointment to serve as an external director, any affiliation with the company to whose board of directors the external director is proposed to be appointed, with any controlling shareholder of the company, with a relative of such controlling shareholder, or with any entity controlled, on the date of such appointment or within the preceding two years, by the company or by a controlling shareholder of the company. If the company has no controlling shareholder or a shareholder holding 25% or more of the company’s voting rights, a person may not serve as an external director if the person has any affiliation, at the time of the appointment, to the chairman of the board of directors, the chief executive officer or the most senior financial officer of the company, or to a shareholder holding 5% or more of the outstanding shares or voting rights of the company.
The term “controlling shareholder” means a shareholder with the ability to direct the activities of the company, other than by virtue of being an office holder. A shareholder is presumed to have “control” of the company and thus to be a controlling shareholder of the company if the shareholder holds 50% or more of the “means of control” of the company. “Means of control” is defined as (1) the right to vote at a general meeting of a company or a corresponding body of another corporation; or (2) the right to appoint directors of the corporation or its general manager.
The term “affiliation” includes:
| ● | an employment relationship; | |
| ● | a business or professional relationship maintained on a regular basis; | |
| ● | or control; and | |
| ● | service as an office holder, excluding service as a director in a private company prior to the first offering of its shares to the public if such director was appointed as a director of the private company in order to serve as an external director following the initial public offering. |
The term “relative” is defined as a spouse, sibling, parent, grandparent, descendant, spouse’s descendant, sibling and parent and the spouse of each of the foregoing.
| 115 |
In addition, no person may serve as an external director if: (i) the person’s other positions or other business activities create, or may create, a conflict of interest with the person’s service as an external director or interfere with the person’s ability to serve as an external director; (ii) at the time such person serves as a non-external director of another company on whose board of directors a director of the reciprocal company serves as an external director; (iii) the person is an employee of the Israel Securities Authority or of an Israeli stock exchange; (iv) such person or such person’s relative, partner, employer or anyone to whom such person is directly or indirectly subordinate, or any entity under such person’s control, has business or professional relations with any person or entity he or she should not be affiliated with, as described above, unless such relations are negligible; or (v) such person received compensation, directly or indirectly, in connection with such person’s services as an external director, other than as permitted under the Companies Law and the regulations promulgated thereunder. If, at the time of election of an external director, all other directors who are not controlling shareholders of such company or their relatives, are of the same gender, then the designated external director must be of the other gender.
Pursuant to the Companies Law, an external director is required to have either financial and accounting expertise or professional qualifications according to criteria set forth in regulations promulgated under the Companies Law, provided that at least one of the external directors has financial and accounting expertise. However, if at least one of the other directors (1) meets the independence requirements of the Exchange Act, (2) meets the Nasdaq requirements for membership on the audit committee and (3) has financial and accounting expertise as defined in the Companies Law and applicable regulations, then neither of our external directors is required to possess financial and accounting expertise as long as both possess other requisite professional qualifications as required under the Companies Law and regulations promulgated thereunder.
In March 2020, our Board adopted the exemption under the Regulation and opted-out from the requirement to have external directors serving on our Board.
Our Board has determined that the minimum number of directors with financial and accounting expertise, in addition to the external director or directors who have such expertise, will be one, and that Mr. Poshinski qualifies as such. In addition, our Board has determined that Mr. Poshinski qualifies as an audit committee financial expert pursuant to the applicable SEC rules, and accordingly as having the necessary financial sophistication as required by the Nasdaq Capital Market rules.
Director Independence
Following our “opt-out” of the requirement to have external directors serving on our Board, we comply with the director independence requirements and the audit committee and the compensation committee composition requirements under U.S. laws (including applicable Nasdaq Capital Market rules) applicable to U.S. domestic issuers. Our Board has undertaken a review of the independence of each director. Based on information provided by each director concerning their background, employment and affiliations, our Board has determined that Mr. Nir, Mr. Poshinski, Dr. Sidransky, Dr. Brosgart and Mr. Heinberg do not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and that each of these directors is “independent” as that term is defined under the listing standards of the Nasdaq. In making these determinations, our Board considered the current and prior relationships that each non-employee director has with our company and all other facts and circumstances our Board deemed relevant in determining their independence, including the beneficial ownership of our capital shares by each non-employee director.
Committees of the Board
Our Articles also provide that the Board may delegate any, or all, of its powers to one or more committees of the Board, and may entrust to and confer upon a “managing director” such of its powers as it deems appropriate. However, the Companies Law provides that certain powers and authorities (for example, the power to approve the financial statements) may not be delegated and may be exercised only by the Board. Notwithstanding the foregoing, we currently do, and intend to continue to, comply with the corporate governance requirements of the Nasdaq Capital Market, except to the extent indicated elsewhere in this annual report, including as set forth under “Item 16G. Corporate Governance” below. The Companies Law requires public companies such as the Company to appoint an audit committee and a remuneration committee.
| 116 |
Audit Committee
The Companies Law requires public companies to appoint an audit committee comprised of at least three directors, including all of the external directors, the majority of whom must be independent directors under the Companies Law. The Companies Law further stipulates that the following may not be members of the audit committee: (i) the chairman of the board of directors; (ii) any director employed by or providing services on an ongoing basis to the company, to a controlling shareholder of the company or an entity controlled by a controlling shareholder of the company; (iii) a director whose livelihood mainly depends on a controlling shareholder; and (iv) a controlling shareholder or any relative of a controlling shareholder.
The Companies Law further requires that: (i) the chairperson of the audit committee must be an external director; (ii) generally, any person who is not entitled to be a member of the audit committee may not attend the audit committee’s meetings and voting sessions, unless such person was invited by the chairperson of the committee for the purpose of presenting a specific subject matter thereof; and (iii) the quorum required for the convening of meetings of the audit committee and for adopting resolutions by the audit committee is a majority of the members of the audit committee, provided that the majority of the members present are independent directors and at least one of them is an external director. As noted, under the Regulation, companies with no controlling shareholder whose shares are listed for trading on specified exchanges outside of Israel, including the Nasdaq Capital Market, may adopt exemptions from various corporate governance requirements of the Companies Law so long as the company satisfies the applicable foreign country laws and regulations, including applicable stock exchange rules, that apply to companies organized in that country relating to the appointment of independent directors and the composition of audit and compensation committees. Such exemptions include an exemption from the requirement to appoint external directors and the requirement that an external director be a member of certain committees. In accordance with these Regulations, we elected to “opt out” from such requirements of the Companies Law.
The responsibilities of the audit committee under the Companies Law include: (i) identifying flaws in the management of a company’s business and making recommendations to the board of directors as to how to correct them; (ii) with respect to certain actions involving conflicts of interest and with respect to certain related party transactions, deciding whether such actions are material actions and whether such transactions are extraordinary transactions, respectively, all for the purpose of approving such actions or transactions; (iii) reviewing and deciding whether to approve certain related party transactions and certain actions involving conflicts of interest; (iv) reviewing the internal auditor’s work program; (v) examining the company’s internal control structure and processes, the performance of the internal auditor and whether the internal auditor has at his or her disposal the tools and resources required to perform his or her duties, considering, inter alia, the special needs of the company and its size; (vi) examining the independent auditor’s scope of work as well as the independent auditor’s fees and providing its recommendations to the appropriate corporate organ; (vii) providing for arrangements as to the manner in which the company will deal with employee complaints with respect to deficiencies in the management of the company’s business and the protection to be provided to such employees; and (viii) with respect to related party transactions with a controlling shareholder, regardless of whether such transactions are extraordinary transactions, that prior to entering into such transaction, to establish the requirement of having a competitive process under the supervision of the audit committee or any individual, committee or body on its behalf and according to criteria established by the audit committee and to determine procedures for approving certain related party transactions with a controlling shareholder, which were determined by the audit committee to be non-extraordinary transactions, but which are not negligible transactions.
| 117 |
Our Board has adopted an audit committee charter setting forth the responsibilities of the audit committee consistent with the rules of the SEC and the Nasdaq Listing Rules, as well as the requirements for such committee under the Companies Law, as described below.
Our audit committee oversees the accounting and financial reporting processes of the Company. It also provides assistance to the Board in fulfilling its legal and fiduciary obligations with respect to matters involving the accounting, auditing, financial reporting and internal control functions of the Company. In carrying out its duties, our audit committee meets with management at least once a quarter, at which time, among other things, it reviews, and either approves or disapproves, the financial results of the Company for the immediately preceding calendar quarter and conveys its conclusions in this regard to the Board. Our audit committee also monitors generally the services provided by the Company’s independent auditors to ensure their independence and reviews all audit and non-audit services provided by them.
Our Board has resolved to delegate to the audit committee the power to pre-approve non-auditing services rendered by the Company’s independent auditors without the need for further approval by our Board. As such, on March 15, 2016, our audit committee approved the adoption of a pre-approval policy, such that the Chairman of the audit committee is authorized to pre-approve any engagement of our independent auditors during a period of twelve months from the date of such approval, for the provision of non-auditing services, for fees not to exceed $20,000, and any such engagement which exceeds $20,000 shall require a pre-approval by the entire audit committee. Once services have been pre-approved, our management must then report to the audit committee on a periodic basis regarding the extent of services actually provided in accordance with the pre-approval policy, and regarding the fees for the services performed.
The Company’s independent and internal auditors also report regularly to our audit committee, and our audit committee discusses with the Company’s independent auditors the quality, not just the acceptability, of the accounting principles, the reasonableness of significant judgments and the clarity of disclosures in the Company’s financial statements, as and when it deems it appropriate to do so.
Under the provisions of the Sarbanes-Oxley Act, the audit committee is directly responsible for the appointment, compensation and oversight of the work of the company’s independent auditors. However, under Israeli law, the appointment of independent auditors and their compensation require the approval of the shareholders of a public company. Pursuant to Israeli law, the shareholders may delegate the authority to determine the compensation of the independent auditors to the board of directors. In addition, pursuant to the Companies Law, the audit committee is required to examine the independent auditors’ fees and to provide its recommendations with respect thereto to the appropriate corporate body. Accordingly, the appointment of our independent auditors is required to be approved and recommended to the shareholders by our audit committee and Board and approved by the shareholders. The compensation of the independent auditors for audit services is required to be approved and recommended to the Board by our audit committee and approved by the Board. The Board has delegated its authority to approve the compensation of independent auditors for non-auditing services to the audit committee.
Mr. Nir, Mr. Poshinski and Dr. Sidransky are the current members of our audit committee, with Mr. Poshinski serving as chairperson. Each of our audit committee members are “independent directors” in accordance with the Nasdaq Capital Market corporate governance requirements, as affirmatively determined by our Board. In addition, our Board has affirmatively determined that Mr. Poshinski also qualifies as an audit committee financial expert pursuant to the applicable SEC rules, and accordingly has the necessary financial sophistication as required by the Nasdaq Capital Market rules.
| 118 |
Remuneration Committee
The Companies Law requires public companies to appoint a remuneration committee comprised of at least three directors, including all of the external directors, who must generally also constitute a majority of the members. All other members of the committee, who are not external directors, must be directors who receive compensation consistent with that of external directors and that is in compliance with the Compensation Regulations. In addition, the chairperson of the remuneration committee must be an external director. As noted, under the Regulation, we elected to “opt out” from such requirements of the Companies Law.
The Companies Law further stipulates that directors who are not qualified to serve on the audit committee, as described above, may not serve on the remuneration committee either and that similar to the audit committee, generally, any person who is not entitled to be a member of the remuneration committee may not attend the remuneration committee’s meetings. Our Board has adopted a remuneration committee charter setting forth the responsibilities of our remuneration committee, as described below.
The responsibilities of the remuneration committee under the Companies Law include: (i) making recommendations to the board of directors with respect to the approval of the compensation policy and any extensions thereto; (ii) periodically reviewing the implementation of the compensation policy and providing the board of directors with recommendations with respect to any amendments or updates thereto; (iii) reviewing and resolving whether or not to approve transactions with respect to the terms of office and employment of Office Holders; and (iv) resolving, under certain circumstances prescribed under the Companies Law, whether or not to exempt a transaction with a candidate for chief executive officer who meets non-affiliation criteria from shareholder approval.
Our remuneration committee also oversees the administration of the Company’s various compensation plans and arrangements, in particular, the incentive compensation, deferred compensation and equity based plans of the Company (and to the extent appropriate, of the subsidiaries of the Company) and assists the Board in fulfilling its responsibilities relating to the compensation of directors, the Chief Executive Officer and other Office Holders of the Company. In carrying out these duties, our remuneration committee meets on an ad hoc basis. Under the Companies Law, our remuneration committee may need to seek the approval of the Board and the shareholders for certain compensation decisions as described above. Each member of our remuneration committee is an “independent director” in accordance with the Nasdaq Capital Market corporate governance requirements, as affirmatively determined by our Board. Mr. Nir, Mr. Poshinksi and Dr. Sidransky are the current members of our remuneration committee, with Dr. Sidransky serving as chairperson.
Compensation Policy
As approved by our shareholders, and as required by the Companies Law, we have adopted a Compensation Policy regarding the terms of office and employment of our “office holders” (as defined under the Companies Law, which includes directors, the CEO, other executive officers and any other managers directly subordinate to the CEO), including cash compensation, equity-based awards, releases from liability, indemnification and insurance, severance and other benefits. Each of the named executive officers is an “office holder” within the meaning of the Companies Law. The Compensation Policy is reviewed from time to time by our remuneration committee and our Board to ensure its appropriateness, and is required to be brought at least once every three years to our shareholders for approval. See “Item 6. Directors, Senior Management and Employees — B. Compensation — Certain Approvals Required for Office Holders’ Compensation of the Companies Law”.
Our most recent Compensation Policy was last approved at our annual general meeting of shareholders that was held on August 13, 2020. The Compensation Policy links pay to performance and aligns our executive officers’ interests with those of the Company and of our shareholders. It allows us to provide meaningful incentives that reflect both our short and long-term goals and performance, as well as the executive officers’ individual performance and impact on shareholder value, while providing compensation that is competitive in the global marketplace in which we recruit talent and is designed to reduce incentives for our executive officers to take excessive risks.
| 119 |
The Compensation Policy emphasizes each executive officer’s individual characteristics (such as his or her respective position, education, scope of responsibilities and contribution to the attainment of our goals) as the basis for compensation variation among executive officers, taking into account the internal ratios between compensation of our executive officers and directors and other employees of the Company. Pursuant to the Compensation Policy, the compensation that may be granted to an executive officer may include: base salary and benefits, annual cash bonuses and other cash bonuses (such as retention and special bonuses), as well as equity-based compensation, retirement and termination of employment benefits and other benefits. The cash bonuses that may be granted under the Compensation Policy are limited to a maximum amount linked to the executive officer’s base salary.
Under the Compensation Policy, an annual cash bonus that will be awarded to executive officers (other than the CEO) will be based on performance objectives and a discretionary evaluation of the executive officer’s overall performance by the CEO and may be subject to minimum thresholds. The remuneration committee and the Board will determine any applicable minimum thresholds that must be met for entitlement to the annual cash bonus (all or any portion thereof) and the formula for calculating any annual cash bonus payout on the basis of, but not limited to, company and individual objectives. Notwithstanding the above, we may determine that, with respect to any executive officer subordinated to the CEO, which does not serve as a director, a portion or all of his or her annual cash bonus will be based on the evaluation of the CEO.
The Compensation Policy provides that the annual bonus awarded to the Company’s CEO will be mainly based on measurable objectives of the Company, subject to a minimum threshold on the basis of, but not limited to, company and personal objectives. 25% or less of the annual cash bonus granted to the Company’s CEO may be based on a discretionary evaluation of the CEO’s overall performance by the remuneration committee and the Board. The measurable objectives will be determined annually by the remuneration committee and the Board at the commencement of each fiscal year, or upon engagement, in case of newly hired CEO, or in other special circumstances as set forth in the Compensation Policy.
The equity-based compensation under the Compensation Policy for our executive officers is designed in a manner intended to attract and retain officers and align their interests with shareholders’ interests to maximize creation of long-term economic value for the Company, and to strengthen the retention and the motivation of executive officers in the long term. Equity-based awards may be granted from time to time in the form of options and/or other equity- based awards, such as RSUs in accordance with our 2013 Plan as may be updated from time to time.
The Compensation Policy contains compensation recovery provisions in the event of accounting restatement, which would allow us, under certain conditions, to recover bonuses or performance-based equity paid in excess of what would have been paid under the financial statements, as restated. The Compensation Policy also contains provisions that allow us to exculpate, indemnify and insure our executive officers and directors subject to certain updated limitations set forth in the Compensation Policy.
Based on information provided to us by our insurance brokers (and which has been supported by our independent insurance consultants), there has recently been a significant increase in the cost of D&O liability insurance for non US companies listed in the US, and especially in the life sciences sector. The increases have been tied to extensive losses suffered by the D&O insurers as a result, among other things, of significant increases in the number of class actions filed against Nasdaq listed companies. For example, the year 2018 set a 20-year record high for securities class actions filed against issuers of common or preferred stock listed in the US. Due to the above mentioned market environment, insurers adopted a very defensive and selective approach and some of the insurers are no longer providing US traded companies with new offers and those that are still active in the market have been increasing their level of compensation (in the form of premiums), which they believe have not been commensurate with the risk being taken by them. In parallel, there has been an increase in the amounts of the deductibles payable by public companies in situations in which an insurable event occurs. As a result, our most recent Compensation Policy reflects an increase of the premiums payable in order to maintain the coverage levels under our renewed D&O insurance policy.
The Compensation Policy also governs the compensation of our board members and provides that our directors will be entitled to an annual cash fee retainer (which shall not exceed 20% of the annual base salary paid to our CEO during the year 2019 plus VAT) and may be paid through the grant of equity awards up to the limits set forth in the Compensation Policy.
| 120 |
Nominating Committee
The Nasdaq Capital Market corporate governance requires each company adopting a nominating committee to certify that it has adopted a formal written charter or board resolution, as applicable, addressing the nominations process and such related matters as may be required under U.S. federal securities laws. Although not required as a foreign private issuer to adopt a nominating committee, we have decided to follow such requirement.
Our Board has adopted a nominating committee charter setting forth the responsibilities of the nominating committee consistent with the Nasdaq Listing Rules.
The nominating committee is responsible for identifying individuals qualified to be appointed as board members, and recommending to the Board appropriate director nominees for election at the general meeting of shareholders.
Independent director oversight of nominations enhances investor confidence in the selection of well-qualified director nominees, as well as independent nominees as required by the rules. The Nasdaq Capital Market listing rule is also intended to provide flexibility for a company to choose an appropriate board structure and reduce resource burdens, while ensuring that independent directors approve all nominations.
Mr. Nir, Mr. Poshinski, and Dr. Sidransky are the current members of our nominating committee, with Dr. Sidransky serving as chairperson. Nasdaq Capital Market Listing Rule 5605(e) requires that our nominating committee be comprised solely of independent directors unless the nominating committee is comprised of at least three members and the Board determines that such non-independent director’s membership, which shall not be longer than two years, is required by the best interests of the Company and our shareholders.
R&D Committee
Our R&D Committee, which was established by the Board on May 2014, advises and assists the Board in its oversight of our research and development programs, including the rationale and timeline of clinical trials and other studies, as well as market surveys in connection therewith. The R&D Committee operates in accordance with the purposes and objectives determined by the Board from time to time. Dr. Sidransky, Dr. Brosgart and Mr. Baharaff are the current members of our R&D Committee, with Dr. Sidransky serving as chairperson.
Internal Auditor
Under the Companies Law, the board of directors of an Israeli public company must appoint an internal auditor recommended by the audit committee and nominated by the board of directors. The role of the internal auditor is to examine, among other things, our compliance with applicable law and orderly business procedures. An internal auditor should comply with the requirements of the Companies Law and the Internal Audit Law, 5752-1992, and may not be:
| (a) | a person (or a relative of a person) who holds more than 5% of the Company’s outstanding shares or voting rights; | |
| (b) | a person (or a relative of a person) who has the power to appoint a director or the general manager of the Company; |
| 121 |
| (c) | an Office Holder, including a director, of the Company (or a relative thereof); or | |
| (d) | a member of the Company’s independent accounting firm, or anyone on his or her behalf. |
Pursuant to Israeli law, an internal auditor’s tenure cannot be terminated without his or her consent, nor can he or she be suspended from such position unless the board of directors of the company has so resolved following the recommendations of the company’s audit committee and, after providing the internal auditor with the opportunity to present his or her position to the board of directors of the company and to the audit committee.
On January 12, 2021, our Board appointed Zach Refaeli, CPA, from Ernst & Young Israel - Kost Forer Gabbay & Kasierer, Tel Aviv, Israel, as the Company’s internal auditor for a period of three years, effective as of January 12, 2021.
Exculpation and Indemnification of Directors and Officers
Under the Companies Law, a company may not exculpate an Office Holder from liability for a breach of the duty of loyalty. An Israeli company may exculpate an Office Holder in advance from liability to the company, in whole or in part, for damages caused to the company as a result of a breach of the duty of care but only if a provision authorizing such exculpation is included in its articles of association. Our Articles include such a provision. The Company may not exculpate in advance a director from liability arising out of a prohibited dividend or distribution to shareholders.
Under the Companies Law, and the Securities Law, 5738—1968, or the Securities Law, a company may indemnify, or undertake in advance to indemnify, an Office Holder for the following liabilities and expenses, imposed on Office Holder or incurred by Office Holder due to acts performed by him or her as an Office Holder, provided its articles of association include a provision authorizing such indemnification:
| ● | a monetary liability incurred by or imposed on him or her in favor of another person pursuant to a judgment, including a settlement or arbitrator’s award approved by a court. However, if an undertaking to indemnify an Office Holder with respect to such liability is provided in advance, then such an undertaking must be limited to events which, in the opinion of the board of directors, can be foreseen based on the company’s activities when the undertaking to indemnify is given, and to an amount or according to criteria determined by the board of directors as reasonable under the circumstances, and such undertaking shall detail the abovementioned foreseen events and amount or criteria; | |
| ● | reasonable litigation expenses, including attorneys’ fees, incurred by the Office Holder as a result of an investigation or proceeding instituted against him or her by an authority authorized to conduct such investigation or proceeding, provided that (i) no indictment was filed against such Office Holder as a result of such investigation or proceeding; and (ii) no financial liability was imposed upon him or her as a substitute for the criminal proceeding as a result of such investigation or proceeding or, if such financial liability was imposed, it was imposed with respect to an offense that does not require proof of criminal intent or as a monetary sanction; | |
| ● | a monetary liability imposed on him or her in favor of an injured party at an Administrative Procedure (as defined below) pursuant to Section 52(54)(a)(1)(a) of the Securities Law; | |
| ● | expenses incurred by an office holder or certain compensation payments made to an injured party that were instituted against an office holder in connection with an Administrative Procedure under the Securities Law, including reasonable litigation expenses and reasonable attorneys’ fees; and |
| 122 |
| ● | reasonable litigation expenses, including attorneys’ fees, incurred by the Office Holder or imposed by a court in proceedings instituted against him or her by the company, on its behalf, or by a third-party, or in connection with criminal proceedings in which the Office Holder was acquitted, or as a result of a conviction for an offense that does not require proof of criminal intent. | |
| ● | An “Administrative Procedure” is defined as a procedure pursuant to chapters H3 (Monetary Sanction by the Israeli Securities Authority), H4 (Administrative Enforcement Procedures of the Administrative Enforcement Committee) or I1 (Arrangement to prevent Procedures or Interruption of procedures subject to conditions) to the Securities Law. |
Under the Companies Law and the Securities Law, a company may insure an Office Holder against the following liabilities incurred for acts performed by him or her as an Office Holder if and to the extent provided in the company’s articles of association:
| ● | a breach of the duty of loyalty to the company, provided that the Office Holder acted in good faith and had a reasonable basis to believe that such act would not prejudice the company; | |
| ● | a breach of the duty of care to the company or to a third-party; | |
| ● | a monetary liability imposed on the Office Holder in favor of a third-party; | |
| ● | a monetary liability imposed on the office holder in favor of an injured party at an Administrative Procedure pursuant to Section 52(54)(a)(1)(a) of the Securities Law; and | |
| ● | expenses incurred by an office holder in connection with an Administrative Procedure instituted against him or her, including reasonable litigation expenses and reasonable attorneys’ fees. |
Nevertheless, under the Companies Law, a company may not indemnify, exculpate or insure an Office Holder against any of the following:
| ● | a breach of the duty of loyalty, except for indemnification and insurance for a breach of the duty of loyalty to the company in the event Office Holder acted in good faith and had a reasonable basis to believe that the act would not prejudice the company; | |
| ● | a breach of the duty of care committed intentionally or recklessly, excluding a breach arising out of the negligent conduct of the Office Holder; | |
| ● | an act or omission committed with intent to derive unlawful personal benefit; or | |
| ● | a fine, monetary sanction, penalty or forfeit levied against the Office Holder. |
Under the Companies Law, exculpation, indemnification and insurance of Office Holders require the approval of the remuneration committee, board of directors and, in certain circumstances, the shareholders, as described above under “Item 6—Directors, Senior Management and Employees—B. Compensation.”
Our Articles permit us to exculpate, indemnify and insure our Office Holders to the fullest extent permitted by the Companies Law. Each of our Office Holders have entered into an indemnification agreement with us, exculpating them, to the fullest extent permitted by Israeli law, from liability to us for damages caused to us as a result of a breach of the duty of care and undertaking to indemnify them to the fullest extent permitted by Israeli law, including with respect to liabilities resulting from certain acts performed by such Office Holders in their capacity as an Office Holder of the Company, our subsidiaries or our affiliates.
| 123 |
In the opinion of the SEC, indemnification of directors and Office Holders for liabilities arising under the Securities Act, however, is against public policy and therefore unenforceable.
Agreements with Directors
Other than a written agreement with our President, Chief Executive Officer and Chairman, as detailed in “Item 6. Directors, Senior Management and Employees—B. Compensation—Employment Agreements and Arrangements with Directors and Related Parties—Employment Agreement with Our President, Chief Executive Officer and Chairman of the Board,” we do not have written agreements with any director providing for benefits upon the termination of his or her services with our Company.
D. Employees.
As of December 31, 2021, we had 24 employees, of which 19 were full-time employees and 5 were part-time employees. 18 of the Company’s employees were involved in our clinical and product development operations and 6 served in general and administrative capacities.
While none of our employees are party to any collective bargaining agreements or represented by any labor unions, certain provisions of the Israeli labor laws and certain collective bargaining agreements between the Histadrut (General Federation of Labor in Israel) and the Coordination Bureau of Economic Organizations (including the Industrialists’ Associations) are applicable to our employees by order of the Israel Ministry of Economics. These provisions primarily concern the length of the workday, minimum daily wages for professional workers, pension fund benefits for all employees, insurance for work-related accidents, procedures for dismissing employees, determination of severance pay and other conditions of employment. We generally provide our employees with benefits and working conditions beyond the required minimums. We have never experienced any employment-related work stoppages and believe our relationship with our employees is favorable.
E. Share Ownership.
The following table sets forth information regarding beneficial ownership of our ordinary shares as of April 15, 2022, the latest practicable date for inclusion in this annual report, held by our directors and executive officers, individually and as a group and beneficial owners of more than 5% of our outstanding shares.
Beneficial ownership is determined in accordance with the rules of the SEC and includes voting or investment power with respect to ordinary shares. Ordinary shares issuable under share options, warrants or other conversion rights currently exercisable or that are exercisable within 60 days after April 15, 2022 are deemed outstanding for the purpose of computing the percentage ownership of the person holding the options, or other conversion rights, but are not deemed outstanding for the purpose of computing the percentage ownership of any other person. Percentage of shares beneficially owned is based on 25,088,414 ordinary shares outstanding on April 15, 2022.
Except as otherwise indicated, and subject to applicable community property laws, the persons named in the table have sole voting and investment power and the right to receive the economic benefit of ownership with respect to all ordinary shares held by that person.
| 124 |
Unless otherwise stated, the address for our directors and senior management is c/o Galmed Pharmaceuticals Ltd., 16 Ze’ev Tiomkin St. Tel Aviv, Israel 6578317.
| Number of ordinary | Percentage of ordinary | |||||||
| shares beneficially | shares beneficially | |||||||
| owned(1) | owned | |||||||
| More than 5% Holders | ||||||||
| BBVF Inc.(2) | 1,799,694 | 7.2 | % | |||||
| Directors and Executive Officers | ||||||||
| Allen Baharaff(3) | 4,468,016 | 17.1 | % | |||||
| Shmuel Nir(4) | 125,646 | * | ||||||
| Dr. David Sidransky(5) | 75,625 | * | ||||||
| Dr. Carol L. Brosgart(6) | 48,125 | * | ||||||
| Marshall Heinberg(7) | 34,779 | * | ||||||
| Dr. Liat Hayardeny(8) | 135,000 | * | ||||||
| Yohai Stenzler(9) | 126,625 | * | ||||||
| Guy Nehemya(10) | 114,125 | * | ||||||
| Amir Poshinski(11) | 17,500 | * | ||||||
| Doron Cohen | 0 | * | ||||||
| All directors and executive officers as a group (10 persons) | 5,147,406 | 19.8 | % | |||||
*Less than 1%.
(1) All options included are either currently exercisable or will be exercisable within 60 days of April 15, 2022.
(2) Based upon information contained in a Statement on Schedule 13G/A filed by the shareholder on February 12, 2021. Shares beneficially owned consist of (i) 923,424 ordinary shares held directly by Biotechnology Value Fund, L.P., or BVF, (ii) 710,895 ordinary shares held directly by Biotechnology Value Fund II, L.P., or BVF2, and (iii) 135,355 ordinary shares held directly by Biotechnology Value Trading Fund OS LP, or Trading Fund OS. BVF I GP LLC, or BVF GP, as the general partner of BVF, may be deemed to beneficially own the 923,424 ordinary shares beneficially owned by BVF. BVF II GP LLC, or BVF2 GP, as the general partner of BVF2, may be deemed to beneficially own the 710,895 ordinary shares beneficially owned by BVF2. BVF Partners OS Ltd. or Partners OS, as the general partner of Trading Fund OS, may be deemed to beneficially own the 135,355 ordinary shares beneficially owned by Trading Fund OS. BVF GP Holdings LLC, or BVF GPH, as the sole member of each of BVF GP and BVF2 GP, may be deemed to beneficially own the 1,634,319 ordinary shares beneficially owned in the aggregate by BVF and BVF2. BVF Partners L.P., or Partners, as the investment manager of BVF, BVF2 and Trading Fund OS, and the sole member of Partners OS, may be deemed to beneficially own the 1,799,694 ordinary shares beneficially owned in the aggregate by BVF, BVF2, Trading Fund OS, and a certain Partners managed account, or the Partners Managed Account, including 30,020 ordinary shares held in the Partners Managed Account. BVF Inc., as the general partner of Partners, may be deemed to beneficially own the 1,799,694 ordinary shares owned by Partners. Mark N. Lampert, as a director and officer of BVF Inc., may be deemed to beneficially own the 1,799,694 ordinary shares beneficially owned by BVF Inc. BVF GP disclaims beneficial ownership of the ordinary shares beneficially owned by BVF. BVF2 GP disclaims beneficial ownership of the ordinary shares beneficially owned by BVF2. Partners OS disclaims beneficial ownership of the Shares beneficially owned by Trading Fund OS. BVF GPH disclaims beneficial ownership of the ordinary shares beneficially owned by BVF and BVF2. Each of Partners, BVF Inc. and Mr. Lampert disclaims beneficial ownership of the ordinary shares beneficially owned by BVF, BVF2, Trading Fund OS, and the Partners Managed Account.
(3) Consists of (i) 3,420,822 ordinary shares, of which 3,416,822 are held through G. Yarom Medical Research Ltd., a company incorporated under the laws of the State of Israel, of which Mr. Baharaff is the controlling shareholder and the chairman of its board of directrs and 4,000 ordinary shares held by Mr. Baharaff, which were purchased in the open market; and (ii) options to purchase 1,047,284 ordinary shares that are currently exercisable within 60 days as of April 15, 2022. Of the 4,413,106 ordinary shares, Mr. Baharaff exercises sole voting and dispositive power over 1,051,284 shares beneficially owned and shared voting and dispositive power with G. Yarom Medical Research Ltd. over 3,416,822 shares.
(4) Consists of (i) 48,938 ordinary shares, of which 41,438 ordinary shares are held through Tushia Consulting Engineers Ltd., of which Shmuel Nir is its controlling shareholder and 7,500 ordinary shares held by Mr. Nir; and (ii) 76,708 ordinary shares issuable upon the exercise of options that are currently exercisable or will be exercisable within 60 days as of April 15, 2022.
| 125 |
(5) Consists of (i) 7,500 ordinary shares held by Dr. Sidransky; and (ii) 68,125 ordinary shares issuable upon the exercise of options that are currently exercisable or will be exercisable within 60 days as of April 15, 2022.
(6) Consists of 48,125 ordinary shares issuable upon the exercise of options that are currently exercisable or will be exercisable within 60 days as of April 15, 2022.
(7) Consists of (i) 8,529 ordinary shares held by Mr. Heinberg; and (ii) 26,250 ordinary shares issuable upon the exercise of options that are currently exercisable or will be exercisable within 60 days as of April 15, 2022.
(8) Consists of 135,000 ordinary shares issuable upon the exercise of options that are currently exercisable or will be exercisable within 60 days as of April 15, 2022.
(9) Consists of (i) 5,625 ordinary shares held by Mr. Stenzler; and (ii) 121,000 ordinary shares issuable upon the exercise of options that are currently exercisable or will be exercisable within 60 days as of April 15, 2022.
(10) Consists of (i) 5,625 ordinary shares held by Mr. Nehemya; and (ii) 108,500 ordinary shares issuable upon the exercise of options that are currently exercisable or will be exercisable within 60 days as of April 15, 2022.
(11) Consists of 17,500 ordinary shares issuable upon the exercise of options that are currently exercisable or will be exercisable within 60 days as of April 15, 2022.
Change in Control
To our knowledge, (i) we are not directly or indirectly owned or controlled by another corporation, by any foreign government or by any other natural or legal person severally or jointly, except as disclosed in the above table regarding our major shareholders, and (ii) there are no arrangements which would result in our change in control at a subsequent date.
Significant Changes in the Ownership of Major Shareholders
To our knowledge, other than as disclosed in the table above, our other filings with the SEC and this annual report, there has been no significant change in the percentage ownership held by any major shareholder since January 1, 2019.
Major Shareholders Voting Rights
Our major shareholders do not have different voting rights.
Record Holders
To our knowledge, as of April 15, 2022, we had one holder of record of our ordinary shares with a U.S. address, Cede & Co., the nominee of The Depository Trust Company. This holder held in the aggregate 21,641,740 ordinary shares, or 86.3% of our outstanding ordinary shares as of April 15, 2022. The number of record holders in the United States is not representative of the number of beneficial holders of our ordinary shares nor is it representative of where such beneficial holders are resident since many of these ordinary shares were held by brokers or other nominees.
| 126 |
2013 Incentive Share Option Plan
We maintain one equity-based incentive plan, our 2013 Incentive Share Option Plan, or the 2013 Plan. As of April 15, 2022, the latest practicable date for inclusion in this annual report, a total of 4,340,492 shares were reserved for issuance under our 2013 Plan, of which (1) options to purchase 2,754,247 ordinary were issued and outstanding thereunder (i.e., were granted but not canceled, expired or exercised); (2) options to purchase 1,091,183 ordinary shares were exercised and 64,028 ordinary shares were issued upon vesting of RSUs; (; and (3) 431,034 shares remain unallocated for future equity awards pursuant to our 2013 Plan.
Our 2013 Plan, which was adopted by our Board on September 2, 2013, and approved by our shareholders in December 30, 2013 (as was amended by the Board and our shareholders on March 30, 2015, May 11, 2015, and August 30, 2018), provides for the grant of options to purchase our ordinary shares and the issuance of RSUs to our officers, directors, employees, service providers and consultants. Our 2013 Plan provides for such equity-based compensation under various and different tax regimes, including those detailed below.
The 2013 Plan is administered by our Board, which, on its own or upon the recommendation of our remuneration committee or any other similar committee of the Board, shall determine, subject to applicable law, the identity of grantees of awards and various terms of the grant. Consistent with our Compensation Policy, the 2013 Plan provides for granting options to purchase our ordinary shares pursuant to Section 102 of the Israeli Income Tax Ordinance, or the Ordinance, under the capital gains route, to directors, officers and employees who are Israeli residents holding (or have a right to hold or to purchase) less than 10% of our total share capital and do not have a right to receive 10% or more of the Company’s profits.
Section 102 of the Ordinance allows Israeli employees, directors and officers, who are not controlling shareholders to receive favorable tax treatment for compensation in the form of shares or options. However, under this route we are not allowed to deduct any expense with respect to the issuance of the options or shares. Israeli non-employee service providers, consultants and shareholders who hold 10% or more of our total share capital or are otherwise controlling shareholders, may be granted options pursuant to Section 3(i) of the Ordinance, which does not provide for similar tax benefits. In order to comply with the terms of the capital gains route pursuant to Section 102 of the Ordinance, the granted options as well as the ordinary shares issued upon exercise of these options and other shares received subsequently following any realization of rights with respect to such options (such as share dividends and share splits), must be granted to a trustee for the benefit of the relevant grantee and should be held by the trustee for at least two years after the date of the grant. If such options or shares are sold by the trustee or are transferred to the grantee before the end of the two-year period, then the grantee would be taxed at top marginal rates upon selling the shares.
For residents, or deemed residents, of the United States, the 2013 Plan provides grants, which are pursuant to Section 422 of the Internal Revenue Code of 1986, as amended, or the Code, as incentive stock options, or ISOs, and any other participants which do not qualify for ISOs, as non-statutory stock options, or NSOs, pursuant to the Code.
Section 422 of the Code allows employees, directors and officers, who are non-controlling shareholders (e.g., less than 10% shareholders) and are considered residents of the United States or those who are deemed to be residents of the United States for purposes of the payment of tax, or are otherwise subject to taxation in the United States with respect to the grant of awards, to receive favorable tax treatment for compensation in the form of shares or ISOs. 10% shareholders or persons which are not service providers will receive NSOs, which do not entitle them to receive similar tax benefits. Section 422(b) of the Code provides for the ISO track such that the individual does not have to pay ordinary income tax (nor employment taxes) on the difference between the exercise price and the fair market value of the shares issued (however, the holder may have to pay U.S. alternative minimum tax instead). However, if the shares are held for one year from the date of exercise and two years from the date of grant, then the profit (if any) made on sale of the shares is taxed as long-term capital gain. Section 422 of the Code requires that any grant of awards shall not be made at a price which is less than 100% of the fair market value of such awards on the date of the grant, all pursuant to the terms of Section 409A of the Code. However, under this ISO track, we are not allowed to deduct any expense with respect to the issuance of the options or shares. In order to comply with the terms of the ISO track, the option granted thereunder must meet the requirements of Section 422 of the Code when granted and at all times until the exercise thereof.
| 127 |
Options and RSUs granted under the 2013 Plan will vest in accordance with the vesting dates as determined by the Board following the recommendation of the remuneration committee or any other similar committee of the Board with respect to each grant. Generally, options that are not exercised within ten years from the grant date expire, unless otherwise determined by the Board and the remuneration committee, as applicable, provided, however, that, pursuant to our Compensation Policy, any equity-based awards to Office Holders must include both a gradual vesting period of at least three years from the date of grant, and an exercise period of no more than ten years from the date of grant.
Upon such date or dates designated in the applicable award agreement, unless earlier forfeited, subject to the receipt of any approvals required from any relevant tax authority, we shall settle each RSU upon vesting by delivering one ordinary share.
In case of termination for reasons of disability or death, the grantee or his legal successor may exercise options that have vested prior to termination within a period of twelve months from the date of disability or death. If we terminate a grantee’s employment or service for cause, all of the grantee’s vested and unvested unexercised options will expire and terminate on the date of termination. If a grantee’s employment or service is terminated for any other reason, the grantee may exercise his or her vested options within 90 days of the date of termination or within a longer period under specified circumstances determined by our Board. Any expired or unvested options shall return to the option pool reserved under the 2013 Plan for reissuance.
In the event of grantee’s termination prior to a vesting date by reason of such grantee’s death or disability, all of such grantee’s RSUs shall immediately become vested as of the date of such termination. In the event of a grantee’s termination for cause prior to settlement, all of such grantee’s RSUs shall immediately be forfeited for no consideration as of the date of such termination. If a grantee’s employment or service is terminated for any other reason, (1) all vesting with respect to such grantee’s RSUs shall cease, (2) all of such grantee’s unvested RSUs shall immediately be forfeited for no consideration as of the date of such termination, and (3) to the extent not already settled, all of such grantee’s vested RSUs shall be settled in accordance with the settlement schedule set forth in the applicable award agreement.
In the event of a merger or consolidation of our company subsequent to which we would no longer exist as a legal entity, or a sale of all, or substantially all, of our ordinary shares or assets or other transaction having a similar effect on us, or a Transaction, any unexercised options then outstanding will be cancelled. Notwithstanding the foregoing, the Board, or the relevant committee of the Board, may determine that the options will not be cancelled but will be assumed or substituted for an appropriate number of the same type of shares or other securities of the successor company as were distributed to the Company or the shareholders in connection with the Transaction. In addition, the Board, or the relevant committee of the Board, may determine to include in certain option agreements either a clause that provides for acceleration of vesting of all or part of the unvested options in the event of a Transaction or the occurrence of another event or a clause which provides that if the optionee’s employment with the successor company is terminated by the successor company without cause within a certain period, not to exceed two years from the closing of such Transaction, all or part of the unvested options shall be accelerated.
| 128 |
Certain Information Concerning Equity Awards to Office Holders
The following tables set forth information, as of April 15, 2022 concerning all outstanding equity awards to Office Holders as of such date.
Options
| Name of | Exercise | Shares subject | Shares | Schedule | ||||||||||||||||
| Office | price per | to the | vested and | Shares | date of | |||||||||||||||
| Holder | Date of grant | share ($) | option | unexercised | unvested | expiration | ||||||||||||||
| Allen Baharaff | December 30, 2013 | NIS0.01 | 83,770 | 83,770 | 0 | Sep-2-2023 | ||||||||||||||
| December 30, 2013 | NIS0.01 | 241,014 | 241,014 | 0 | Sep-2-2023 | |||||||||||||||
| February 4, 2016 | $ | 5.49 | 140,000 | 140,000 | 0 | Feb-04-2026 | ||||||||||||||
| February 4, 2016 | $ | 5.94 | 170,000 | 170,000 | 0 | Feb-04-2026 | ||||||||||||||
| July 10, 2018 | $ | 11.56 | 220,000 | 206,250 | 13,750 | Jul-10-2028 | ||||||||||||||
| December 17, 2019 | $ | 5.12 | 220,000 | 123,750 | 96,250 | Dec-17-2029 | ||||||||||||||
| November 10, 2020 | $ | 3.10 | 220,000 | 68,750 | 151,250 | Nov-17-2030 | ||||||||||||||
| Shmuel Nir | February 21, 2014 | $ | 3.57 | 8,583 | 8,583 | 0 | Sep-02-2023 | |||||||||||||
| May 11, 2015 | $ | 5.49 | 10,000 | 10,000 | 0 | May-11-2025 | ||||||||||||||
| February 4, 2016 | $ | 5.94 | 30,000 | 30,000 | — | Feb-04-2026 | ||||||||||||||
| July 10, 2018 | $ | 11.56 | 30,000 | 28,125 | 1,875 | Jul-10-2028 | ||||||||||||||
| July 15, 2021 | $ | 3.10 | 20,000 | 0 | 20,000 | Jul-10-2031 | ||||||||||||||
| Amir Poshinski | August 13, 2020 | $ | 4.77 | 30,000 | 15,000 | 15,000 | August 13, 2030 | |||||||||||||
| July 15, 2021 | $ | 3.10 | 20,000 | 0 | 20,000 | Jul-10-2031 | ||||||||||||||
| David Sidransky | May 11, 2015 | $ | 5.49 | 10,000 | 10,000 | 0 | May-11-2025 | |||||||||||||
| February 4, 2016 | $ | 5.94 | 30,000 | 30,000 | — | Feb-04-2026 | ||||||||||||||
| July 10, 2018 | $ | 11.56 | 30,000 | 28,125 | 1,875 | Jul-10-2028 | ||||||||||||||
| July 15, 2021 | $ | 3.10 | 20,000 | 0 | 20,000 | Jul-10-2031 | ||||||||||||||
| Dr. Liat Hayardeny | September 6, 2016 | $ | 4.05 | 32,500 | 32,500 | 0 | Sep-06-2026 | |||||||||||||
| January 31, 2017 | $ | 3.84 | 27,500 | 27,500 | 0 | Jan-31-2026 | ||||||||||||||
| July 10, 2018 | $ | 11.56 | 40,000 | 37,500 | 2,500 | Jul-10-2028 | ||||||||||||||
| December 17, 2019 | $ | 5.12 | 40,000 | 22,500 | 17,500 | Dec-17-2029 | ||||||||||||||
| November 10, 2020 | $ | 3.33 | 40,000 | 12,500 | 27,500 | Nov-10-2030 | ||||||||||||||
| Yohai Stenzler | December 30, 2014 | $ | 5.49 | 3,500 | 3,500 | 0 | Dec-30-2024 | |||||||||||||
| January 3, 2016 | $ | 7.61 | 22,500 | 22,500 | 0 | Jan-03-2026 | ||||||||||||||
| November 7, 2017 | $ | 7.48 | 20,000 | 20,000 | 0 | Nov-07-2020 | ||||||||||||||
| July 10, 2018 | $ | 11.56 | 40,000 | 37,500 | 2,500 | Jul-10-2028 | ||||||||||||||
| December 17, 2019 | $ | 5.12 | 40,000 | 22,500 | 17,500 | Dec-17-2029 | ||||||||||||||
| November 10, 2020 | $ | 3.33 | 40,000 | 12,500 | 27,500 | Nov-10-2030 | ||||||||||||||
| Guy Nehemya | December 30, 2014 | $ | 5.49 | 11,000 | 11,000 | 0 | Dec-30-2024 | |||||||||||||
| January 3, 2016 | $ | 7.61 | 22,500 | 22,500 | 0 | Jan-03-2026 | ||||||||||||||
| July 10, 2018 | $ | 11.56 | 40,000 | 37,500 | 2,500 | Jul-10-2028 | ||||||||||||||
| December 17, 2019 | $ | 5.12 | 40,000 | 22,500 | 17,500 | Dec-17-2029 | ||||||||||||||
| November 10, 2020 | $ | 3.33 | 40,000 | 12,500 | 27,500 | Nov-10-2030 | ||||||||||||||
| Carol L. Brosgart | April 25, 2017 | $ | 4.87 | 20,000 | 20,000 | 0 | Apr-25-2027 | |||||||||||||
| July 10, 2018 | $ | 11.56 | 30,000 | 28,125 | 1,875 | Jul-10-2028 | ||||||||||||||
| July 15, 2021 | $ | 3.10 | 20,000 | 0 | 20,000 | Jul-10-2031 | ||||||||||||||
| Marshall Heinberg | November 1, 2018 | $ | 8.95 | 30,000 | 24,3750 | 5,6275 | Nov-1-2028 | |||||||||||||
| July 15, 2021 | $ | 3.10 | 20,000 | 0 | 20,000 | Jul-10-2031 | ||||||||||||||
| Doron Cohen | February 23, 2022 | $ | 1.61 | 40,000 | 0 | 40,000 | Feb-23-2032 | |||||||||||||
| 129 |
RSUs
| Name of | Shares | |||||||||||||
| Office | subject to | Shares | Shares | |||||||||||
| Holder | Date of grant | the RSUs | vested | unvested | ||||||||||
| Shmuel Nir | Feb-04-2016 | 7,500 | 7,500 | 0 | ||||||||||
| David Sidransky | Feb-04-2016 | 7,500 | 7,500 | 0 | ||||||||||
| Yohai Stenzler | Jan-03-2016 | 5,625 | 5,625 | 0 | ||||||||||
| Guy Nehemya | Jan-03-2016 | 5,625 | 5,625 | 0 | ||||||||||
ITEM 7. Major Shareholders and Related Party Transactions.
A. Major Shareholders.
Except as set forth in “Item 6. Directors, Senior Management and Employees—E. Share Ownership,” to the best of our knowledge, no other person who we know beneficially owns 5% or more of the Company’s ordinary shares outstanding as of April 15, 2022, the latest practicable date for inclusion in this annual report. None of our shareholders has different voting rights from other shareholders. Other than as described herein, to the best of our knowledge, we are not owned or controlled, directly or indirectly, by another corporation, by any foreign government or by any natural person or legal persons, severally or jointly, and we are not aware of any arrangement that may, at a subsequent date, result in a change of control of our company.
B. Related Party Transactions.
The following is a summary description of the material terms of those transactions with related parties to which we, or our subsidiaries, are party and which were in effect since January 1, 2021.
Financing Agreement with GRD
We have provided financing to GRD from time to time, pursuant to which the Company and GRD have executed several capital notes for an aggregate outstanding principal amount of approximately $130.5 million. The par value of such notes is in NIS, and they bear no interest nor repayment date; provided, however, that no repayment shall be made before the fifth anniversary from the issuance date of each note.
Agreements with Directors and Officers
Employment and Consulting Agreements. We have entered into written employment or consulting agreements with certain of our Office Holders. These agreements provide for notice periods of varying duration for termination of the agreement by us or by the relevant Office Holder, during which time the Office Holder will continue to receive base salary and benefits. We have also entered into customary non-competition, confidentiality of information and ownership of inventions arrangements with these Office Holders. However, the enforceability of the noncompetition provisions may be limited under applicable law.
| 130 |
Options. Since our inception, we have granted options to purchase our ordinary shares to certain of our Office Holders. Such option agreements may contain acceleration provisions upon certain merger, acquisition, or change of control transactions. See also “Item 6. Directors, Senior Management and Employees—E. Share Ownership”. We describe our 2013 Plan under “Item 6. Directors, Senior Management and Employees—B. Compensation—2013 Incentive Share Option Plan.” If the relationship between us and an Office Holder is terminated except for “cause” (as defined in the 2013 Plan and/or the applicable option award agreement), options that are vested will generally remain exercisable for 90 days after such termination; provided, however, that prior to the date of such termination, our remuneration committee may authorize an extension of the terms of all or part of the vested options beyond the date of such termination for a period not to exceed the period during which the options by their terms would otherwise have been exercisable, and provided further that the vested options may lose their status as incentive stock options and/or approved 102 options if such extension extends beyond the maximum extension authorized by the Ordinance or the Code, as applicable.
RSUs. We have granted RSUs to certain of our Office Holders. Such award agreements may contain acceleration provisions upon certain merger, acquisition, or change of control transactions. See also “Item 6. Directors, Senior Management and Employees—E. Share Ownership.” We describe our 2013 Plan under “Item 6. Directors, Senior Management and Employees—B. Compensation—2013 Incentive Share Option Plan.” If the relationship between us and an Office Holder is terminated, RSUs that are vested shall be settled in accordance with the settlement schedule set forth in the applicable award agreement.
C. Interests of Experts and Counsel.
Not applicable.
ITEM 8. Financial Information.
A. Consolidated Financial Statements and Other Financial Information.
See “Item 18. Financial Statements” for a list of all financial statements filed as part of this annual report.
Legal Matters
We are neither party to any legal or arbitration proceedings, including those relating to bankruptcy, receivership or similar proceedings and those involving any third-party, nor any governmental proceedings pending or known to be contemplated, which may have, or have had in the recent past, significant effects on the Company’s financial position or profitability.
Dividend Policy
We have never declared or paid any cash dividends on our ordinary shares and do not anticipate paying any cash dividends in the foreseeable future. Payment of cash dividends, if any, in the future will be at the discretion of our Board and will depend on then-existing conditions, including our financial condition, operating results, contractual restrictions, capital requirements, business prospects and other factors our Board may deem relevant.
Payment of dividends may also be subject to Israeli withholding taxes. See “Item 10. Additional Information—E. Taxation—Certain Israeli Tax Considerations” for additional information.
| 131 |
B. Significant Changes.
Other than as otherwise described in this Annual Report on Form 20-F and as set forth below, no significant change has occurred in our operations since the date of our consolidated financial statements included in this Annual Report on Form 20-F.
ITEM 9. The Offer and Listing.
A. Offer and Listing Details
Our ordinary shares have been listed on the Nasdaq Capital Market under the symbol “GLMD” since March 13, 2014. Prior to that date, there was no public trading market for our ordinary shares.
B. Plan of Distribution
Not applicable.
C. Market for Ordinary Shares
Our ordinary shares have been quoted on the NASDAQ Capital Market since March 18, 2014 under the symbol “GLMD.”
D. Selling Shareholders
Not applicable.
E. Dilution
Not applicable.
F. Expenses of the issue
Not applicable.
ITEM 10. Additional Information.
A. Share Capital.
Not applicable.
B. Memorandum and Articles of Association.
Our registration number is 51-495351-2. At the 2014 annual general meeting of shareholders, our shareholders adopted our Articles, which became effective on the consummation of our initial public offering in the United States in March 2014. Under Section 2 of our Articles, the purpose of the Company is to engage in any lawful activity.
| 132 |
The following description of our share capital and provisions of our Articles are summaries and do not purport to be complete and are qualified in their entirety by the complete text of the Articles, which are filed as exhibits to this annual report and incorporated by reference herein, and by Israeli law.
Election of Directors
Our Board consists of three classes of directors, with one class being elected each year by shareholders at the Company’s annual general meeting for a term of approximately three years. In accordance with our Articles, directors so elected cannot be removed from office by the shareholders until the expiration of their term of office. Ordinary shares do not have cumulative voting rights. As a result, the holders of ordinary shares that represent a simple majority of the voting power represented at a shareholders’ meeting and voting at the meeting have the power to elect all of the directors put forward for election. For further information as to these appointments, see “Item 6—Directors, Senior Management and Employees—C. Board Practices.”
Under our Articles, a director shall vacate his or her office if that director dies; is declared bankrupt; is declared to be legally incompetent; resigns such office by notice in writing given to the Company; is not re-elected by the shareholders upon expiration of his or her term at the relevant annual general meeting of shareholders; or otherwise as provided in the Companies Law.
Our Articles provide that a director may, by written notice to the Company, appoint another person to serve as an alternate director provided that such appointment is approved by a majority of the directors then in office, and that such appointing director may remove such alternate director. Any alternate director shall be entitled to notice of meetings of the Board and of relevant committees and to attend and vote accordingly, except that the alternate has no standing at any meeting at which the appointing director is present or at which the appointing director is not entitled to participate as provided in the Companies Law. A person who is not qualified to be appointed as a director, or a person who already serves as a director or an alternate director, may not be appointed as an alternate director.
Unless the appointing director limits the time or scope of the appointment, the appointment is effective for all purposes until the earlier of (i) the appointing director ceasing to be a director; (ii) the appointing director terminating the appointment; or (iii) the occurrence, with respect to the alternate, of any of the circumstances under which a director shall vacate his or her office. The appointment of an alternate director does not in itself diminish the responsibility of the appointing director as a director. An alternate director is solely responsible for his or her actions and omissions and is not deemed an agent of the appointing director. See “Item 6—Directors, Senior Management and Employees—C. Board Practices.” At present, there are no effective appointments of alternate directors for our Board.
Borrowing Powers
Our Board may from time to time, and at its reasonable discretion, borrow or secure the payment of any sum or sums of money for reasonable Company purposes. The directors may raise or secure the repayment of such sum or sums in such manner, at such times and upon such terms and conditions in all respects as they see fit and, in particular, by issuing bonds, perpetual or redeemable debentures, debenture stock or any mortgages, charges or other securities on the undertaking of the whole or any part of the property of the Company, both present and future, including current uncalled capital and called but unpaid capital.
For discussions relating to certain compensation-related requirements of the Companies Law, external directors and financial experts, committees of the Board, and exculpation and indemnification of directors and officers, see “Item 6 - Directors, Senior Management and Employees.”
Fiduciary Duties of Directors and Executive Officers
The Companies Law codifies the fiduciary duties that Office Holders owe to a company. Each person listed in the table under “Item 6. Directors, Senior Management and Employees—A. Directors and Senior Management” is an Office Holder under the Companies Law.
| 133 |
An Office Holder’s fiduciary duties consist of a duty of care and a duty of loyalty. The duty of care requires an Office Holder to act with the level of care with which a reasonable Office Holder in the same position would have acted under the same circumstances. The duty of loyalty requires that an Office Holder act in good faith and in the best interests of a company. The duty of care includes a duty to use reasonable means to obtain:
| ● | information on the advisability of a given action brought for his or her approval or performed by virtue of his or her position; and | |
| ● | all other important information pertaining to these actions. |
The duty of loyalty requires an Office Holder to act in good faith and for the benefit of a company, and includes a duty to:
| ● | refrain from any conflict of interest between the performance of his or her duties to the company and his or her other duties or personal affairs; | |
| ● | refrain from any activity that is competitive with the company; | |
| ● | refrain from exploiting any business opportunity of the company to receive a personal gain for himself or herself or others; and | |
| ● | disclose to the company any information or documents relating to the company’s affairs which the Office Holder received as a result of his or her position as an Office Holder. |
Disclosure of Personal Interests of an Office Holder
The Companies Law requires that an Office Holder promptly disclose to the board of directors any personal interest that he or she may have concerning any existing or proposed transaction with a company, as well as any substantial information or document with respect thereof. An interested Office Holder’s disclosure must be made promptly and, in any event, no later than the first meeting of the board of directors at which the transaction is considered.
Under the Companies Law, a “personal interest” includes an interest of any person in an action or transaction of a company, including a personal interest of one’s relative or of a corporate body in which such person or a relative of such person is a 5% or greater shareholder, director or general manager or in which he or she has the right to appoint at least one director or the general manager, but excluding a personal interest stemming from one’s ownership of shares in a company. A personal interest furthermore includes the personal interest of a person for whom the Office Holder holds a voting proxy or the interest of the Office Holder with respect to his or her vote on behalf of the shareholder for whom he or she holds a proxy, even if such shareholder itself has no personal interest in the approval of the matter. An Office Holder is not, however, obliged to disclose a personal interest if it derives solely from the personal interest of a relative of such Office Holder in a transaction that is not considered an extraordinary transaction.
Under the Companies Law, an extraordinary transaction is defined as any of the following:
| ● | a transaction other than in the ordinary course of business; | |
| ● | a transaction that is not on market terms; or | |
| ● | a transaction that may have a material impact on a company’s profitability, assets or liabilities. |
| 134 |
Approval Procedure
If an Office Holder has a personal interest in a transaction, approval by the board of directors is required for the transaction, unless the articles of association of a company provide for a different method of approval. Our Articles do not provide for any such different method of approval. Further, so long as an Office Holder has disclosed his or her personal interest in a transaction, the board of directors may approve an action by the Office Holder that would otherwise be deemed a breach of the duty of loyalty. However, a company may not approve a transaction or action that is adverse to such company’s interest or that is not performed by the Office Holder in good faith. Approval first by a company’s audit committee and subsequently by the board of directors is required for an extraordinary transaction in which an Office Holder has a personal interest. Arrangements regarding the Office Holders’ terms of office and employment (which includes compensation, indemnification or insurance) generally require the approval of the remuneration committee, board of directors and, in certain circumstances, the shareholders, in that order, and must generally be consistent with the Company’s Compensation Policy, as described under see “Item 6—Directors, Senior Management and Employees—B. Compensation.”
Generally, a person who has a personal interest in a matter which is considered at a meeting of the board of directors or the audit committee may not be present at such a meeting or vote on that matter unless a majority of the directors or members of the audit committee have a personal interest in the matter, or unless the chairman of the audit committee or board of directors (as applicable) determines that he or she should be present in order to present the transaction that is subject to approval. Generally, if a majority of the members of the audit committee and the board of directors (as applicable) has a personal interest in the approval of a transaction, then all directors may participate in discussions of the audit committee and/or the board of directors on such transaction and the voting on approval thereof, but shareholder approval is also required for such transaction.
Transactions with Controlling Shareholders
Pursuant to Israeli law, the disclosure requirements regarding personal interests that apply to directors and executive officers also apply to a controlling shareholder of a public company. In the context of a transaction involving a controlling shareholder or an officer who is a controlling shareholder of a company, a controlling shareholder also includes any shareholder who holds 25% or more of the voting rights if no other shareholder holds more than 50% of the voting rights. Two or more shareholders with a personal interest in the approval of the same transaction are deemed to be a single shareholder and may be deemed a controlling shareholder for the purpose of approving such transaction.
Extraordinary Transactions, including private placement transactions, with a controlling shareholder or in which a controlling shareholder has a personal interest, and engagements with a controlling shareholder or his or her relative, directly or indirectly, including through a corporation under his or her control, regarding the company’s receipt of services from the controlling shareholder, and if such controlling shareholder is also an office holder or an employee of the company, regarding his or her terms of service or employment, require the approval of the audit committee or remuneration committee, the board of directors and the shareholders of a company by a Special Majority, in that order.
Arrangements regarding the terms of office and employment of a controlling shareholder who is an Office Holder, and the terms of employment of a controlling shareholder who is an employee of a company, require the approval of the remuneration committee, board of directors and the shareholders by a Special Majority, in that order, as further described above under “Item 6—Directors, Senior Management and Employees—B. Compensation” with respect to Office Holders’ compensation.
To the extent that any such transaction with a controlling shareholder is for a period extending beyond three years, approval is required once every three years, unless, with respect to extraordinary transactions with a controlling shareholder or in which a controlling shareholder has a personal interest, the audit committee determines that the duration of the transaction is reasonable given the circumstances related thereto.
| 135 |
Dividends and Dividend Policy
Dividends may be distributed only out of profits available for dividends as determined by the Companies Law, provided that there is no reasonable concern that the distribution will prevent the Company from being able to meet its existing and anticipated obligations when they become due. Under the Companies Law, the distribution amount is further limited to the greater of retained earnings or earnings generated over the two most recent years legally available for distribution. In the event that we do not have retained earnings or earnings generated over the two most recent years legally available for distribution, we may seek the approval of the court in order to distribute a dividend. The court may approve our request if it is convinced that there is no reasonable concern that the payment of a dividend will prevent us from satisfying our existing and foreseeable obligations as they become due.
Generally, under the Companies Law, the decision to distribute dividends and the amount to be distributed is made by a company’s board of directors. The Articles provide that the Board may from time to time declare, and cause the Company to pay, such dividends as may appear to it to be justified by the profits of the Company and that the Board has the authority to determine the time for payment of such dividends and the record date for determining the shareholders entitled to receive such dividends, provided the date is not before the date of the resolution to distribute the dividend. Declaration of dividends does not require shareholder approval.
Pursuant to our Articles, subject to the rights of holders of shares with limited or preferred rights, ordinary shares shall confer upon the holders thereof equal rights to receive dividends and to participate in the distribution of the assets of the Company upon its winding-up, in proportion to the amount paid up or credited as paid up on account of the nominal value of the shares held by them respectively and in respect of which such dividends are being paid or such distribution is being made, without regard to any premium paid in excess of the nominal value, if any.
We have never declared or paid any cash dividends on our ordinary shares and do not anticipate paying any cash dividends in the foreseeable future. Payment of cash dividends, if any, in the future will be at the discretion of our Board and will depend on then-existing conditions, including our financial condition, operating results, contractual restrictions, capital requirements, business prospects and other factors our Board may deem relevant.
Payment of dividends may also be subject to Israeli withholding taxes. See “Taxation — Israeli Tax Considerations” for additional information.
Transfer of Shares
Ordinary shares which have been fully paid-up are transferable by submission of a proper instrument of transfer to the Company or its transfer agent together with the certificate of the shares to be transferred and such other evidence, if any, as the directors may require to prove the rights of the intending transferor in the transferred shares.
Our ordinary shares that are fully paid for are issued in registered form and may be freely transferred under our Articles, unless the transfer is restricted or prohibited by applicable law or the rules of a stock exchange on which the shares are traded. The ownership or voting of our ordinary shares by non-residents of Israel is not restricted in any way by our Articles or the laws of the State of Israel, except for ownership by nationals of some countries that are, or have been, declared as enemies of Israel.
Shareholder Meetings
Our Articles provide that an annual general meeting must be held at least once in every calendar year, not later than 15 months after the last preceding annual general meeting, at such time and place as may be determined by the Board. The Board may, in its discretion, convene additional shareholder meetings and, pursuant to the Companies Law, must convene a meeting upon the demand of two directors or one quarter of the directors then in office or upon the demand of the holder or holders of 5% of the Company’s issued share capital and 1% of its voting rights or upon the demand of the holder or holders of 5% of its voting rights. All demands for shareholder meetings must set forth the items to be considered at that meeting. Pursuant to the Companies Law, the holder or holders of 1% of the Company’s voting rights may request the inclusion of an item on the agenda of a future shareholder meeting, provided the item is appropriate for discussion at a shareholder meeting.
| 136 |
The agenda for a shareholder meeting is determined by the Board and must include matters in respect of which the convening of a shareholder meeting was demanded and any matter requested to be included by holder(s) of 1% of the Company’s voting rights. According to regulations promulgated pursuant to the Companies Law and governing the terms of notice and publication of shareholder meetings of public companies, or the General Meeting Regulations, holder(s) of one percent or more of the Company’s voting rights may propose any matter appropriate for deliberation at a shareholder meeting to be included on the agenda of a shareholder meeting, generally by submitting a proposal within seven days of publicizing the convening of a shareholder meeting, or, if the Company publishes a preliminary notice at least 21 days prior to publicizing the convening of a meeting (stating its intention to convene such meeting and the agenda thereof), within 14 days of such preliminary notice. Any such proposal must further comply with the information requirements under applicable law and the Articles.
Pursuant to the Companies Law and regulations promulgated thereunder with respect to the convening of general meetings in a public company, shareholder meetings generally require prior notice of not less than 21 days, and for certain matters specified in the Companies Law, not less than 35 days. The function of the annual general meeting is to elect directors in accordance with the Articles, receive and consider the profit and loss account, the balance sheet and the ordinary reports and accounts of the directors and auditors, appoint auditors and fix their remuneration and transact any other business which under the Articles or applicable law may be transacted by the shareholders of a company in general meeting.
Our Articles determine that the quorum required for either an annual (regular) or an extraordinary (special) general meeting of shareholders consists of at least two shareholders present in person or by proxy holding shares comprising in the aggregate more than 33.33% of the voting rights of the Company. If a meeting is convened by the Board upon the demand of shareholders or upon the demand of less than 50% of the directors then in office or directly by such shareholders or directors and no quorum is present within half an hour from the time appointed, it shall be cancelled. If a meeting is otherwise called and no quorum is present within such time, the meeting is adjourned to the same day one week later at the same time and place or at such other time and place as the Board may determine and specify in the notice of the general meeting and it shall not be necessary to give notice of such adjournment. If a quorum is not present within half an hour from the time stated for such adjourned meeting, any two shareholders present in person or by proxy at such meeting shall constitute a quorum even if, between them, they represent shares conferring 33.33% or less of the voting rights of the Company.
Generally, under the Companies Law and the Articles, shareholder resolutions are deemed adopted if approved by the holders of a simple majority of the voting rights represented at a meeting and voting unless a different majority is required by law or pursuant to the Articles. The Companies Law provides that resolutions on certain matters, such as amending a company’s articles of association, assuming the authority of the board of directors in certain circumstances, appointing auditors, appointing external directors (if applicable), approving certain transactions, increasing or decreasing the registered share capital and approving most mergers must be made by the shareholders at a general meeting. A company may determine in its articles of association certain additional matters in respect of which resolutions by the shareholders in a general meeting will be required.
Access to Corporate Records
Under the Companies Law, all shareholders generally have the right to review minutes of our general meetings, our shareholder register and register of significant shareholders (as defined in the Companies Law), our articles of association, our financial statements, other documents as provided in the Companies Law, and any document we are required by law to file publicly with the Israeli Companies Registrar. Any shareholder who specifies the purpose of its request may request to review any document in our possession that relates to: (i) any action or transaction with a related party which requires shareholder approval under the Companies Law; or (ii) the approval, by the board of directors, of an action in which an office holder has a personal interest. We may deny a request to review a document if we determine that the request was not made in good faith, or if such denial is necessary to protect our interest or protect a trade secret or patent.
| 137 |
Shareholder Duties
Pursuant to the Companies Law, a shareholder has a duty to act in good faith and in a customary manner toward a company and other shareholders and to refrain from abusing his or her power in the company, including, among other things, in voting at the general meeting of shareholders and at class shareholder meetings with respect to the following matters:
| ● | an amendment to the company’s articles of association; | |
| ● | an increase of the company’s authorized share capital; | |
| ● | a merger; or | |
| ● | approval of interested party transactions and acts of Office Holders that require shareholder approval. |
In addition, a shareholder also has a general duty to refrain from discriminating against other shareholders.
Certain shareholders have a further duty of fairness toward a company. These shareholders include any controlling shareholder, any shareholder who knows that it has the power to determine the outcome of a shareholder vote or a shareholder class vote and any shareholder who has the power to appoint or to prevent the appointment of an Office Holder of the company or other power towards the company. The Companies Law does not define the substance of this duty of fairness, except to state that the remedies generally available upon a breach of contract will also apply in the event of a breach of the duty to act with fairness, taking the shareholder’s position in the company into account.
Mergers and Acquisitions under Israeli Law
(i) Merger
The Companies Law permits merger transactions if approved by each party’s board of directors, and, unless certain requirements described under the Companies Law are met, a majority of each party’s shareholders, by a majority of each party’s shares that are voted on the proposed merger at a shareholders’ meeting.
The board of directors of a merging company is required pursuant to the Companies Law to discuss and determine whether in its opinion there exists a reasonable concern that as a result of a proposed merger, the surviving company will not be able to satisfy its obligations towards its creditors, taking into account the financial condition of the merging companies. If the board of directors has determined that such a concern exists, it may not approve a proposed merger. Following the approval of the board of directors of each of the merging companies, the boards of directors must jointly prepare a merger proposal for submission to the Israeli Registrar of Companies.
For purposes of the shareholder vote, unless a court rules otherwise, the merger will not be deemed approved if a majority of the shares voting at the shareholders meeting (excluding abstentions) that are held by parties other than the other party to the merger, any person who holds 25% or more of the means of control of the other party to the merger or any one on their behalf including their relatives or corporations controlled by any of them, vote against the merger. In addition, if the non-surviving entity of the merger has more than one class of shares, the merger must be approved by each class of shareholders.
| 138 |
If the transaction would have been approved but for the separate approval of each class of shares or the exclusion of the votes of certain shareholders as provided above, a court may still rule that the company has approved the merger upon the request of holders of at least 25% of the voting rights of a company, if the court holds that the merger is fair and reasonable, taking into account the appraisal of the merging companies’ value and the consideration offered to the shareholders.
Under the Companies Law, each merging company must send a copy of the proposed merger plan to its secured creditors. Unsecured creditors are entitled to receive notice of the merger, as provided by the regulations promulgated under the Companies Law. Upon the request of a creditor of either party to the proposed merger, the court may delay or prevent the merger if it concludes that there exists a reasonable concern that, as a result of the merger, the surviving company will be unable to satisfy the obligations of the target company. The court may also give instructions in order to secure the rights of creditors.
In addition, a merger may not be completed unless at least 50 days have passed from the date that a proposal for approval of the merger was filed with the Israeli Registrar of Companies and 30 days from the date that shareholder approval of both merging companies was obtained.
(ii) Special Tender Offer
The Companies Law provides that an acquisition of shares of an Israeli public company must be made by means of a special tender offer if as a result of the acquisition the purchaser would become a holder of 25% or more of the voting rights in the company. This rule does not apply if there is already another holder of 25% or more of the voting rights in the company. Similarly, the Companies Law provides that an acquisition of shares in a public company must be made by means of a special tender offer if as a result of the acquisition the purchaser would become a holder of more than 45% of the voting rights in the company, if there is no other shareholder of the company who holds more than 45% of the voting rights in the company.
These requirements do not apply if the acquisition (i) occurs in the context of a private offering, on the condition that the shareholders’ meeting approved the acquisition as a private offering whose purpose is to give the acquirer at least 25% of the voting rights in the company if there is no person who holds at least 25% of the voting rights in the company, or as a private offering whose purpose is to give the acquirer 45% of the voting rights in the company, if there is no person who holds 45% of the voting rights in the company; (ii) was from a shareholder holding at least 25% of the voting rights in the company and resulted in the acquirer becoming a holder of at least 25% of the voting rights in the company; or (iii) was from a holder of more than 45% of the voting rights in the company and resulted in the acquirer becoming a holder of more than 45% of the voting rights in the company.
The special tender offer may be consummated only if (i) at least 5% of the voting power attached to the company’s outstanding shares will be acquired by the offeror and (ii) the special tender offer is accepted by a majority of the votes of those offerees who gave notice of their position in respect of the offer; in counting the votes of offerees, the votes of a holder of control in the offeror, a person who has personal interest in acceptance of the special tender offer, a holder of at least 25% of the voting rights in the company, or any person acting on their or on the offeror’s behalf, including their relatives or companies under their control, are not taken into account.
In the event that a special tender offer is made, a company’s board of directors is required to express its opinion on the advisability of the offer or shall abstain from expressing any opinion if it is unable to do so, provided that it gives the reasons for its abstention. In addition, the board of directors must disclose any personal interest each of member of the board of directors have in the offer or stems therefrom.
An office holder in a target company who, in his or her capacity as an office holder, performs an action the purpose of which is to cause the failure of an existing or foreseeable special tender offer or is to impair the chances of its acceptance, is liable to the potential purchaser and shareholders for damages resulting from his acts, unless such office holder acted in good faith and had reasonable grounds to believe he or she was acting for the benefit of the company. However, office holders of the target company may negotiate with the potential purchaser in order to improve the terms of the special tender offer, and may further negotiate with third parties in order to obtain a competing offer.
| 139 |
If a special tender offer was accepted by a majority of the shareholders who announced their stand on such offer, then shareholders who did not respond to the special offer or had objected to the special tender offer may accept the offer within four days of the last day set for the acceptance of the offer. In the event that a special tender offer is accepted, then the purchaser or any person or entity controlling it and any corporation controlled by them shall refrain from making a subsequent tender offer for the purchase of shares of the target company and may not execute a merger with the target company for a period of one year from the date of the offer, unless the purchaser or such person or entity undertook to effect such an offer or merger in the initial special tender offer.
(iii) Full Tender Offer
Under the Companies Law, a person may not acquire shares in a public company if, after the acquisition, he will hold more than 90% of the shares or more than 90% of any class of shares of that company, unless a tender offer is made to purchase all of the shares or all of the shares of the particular class. The Companies Law also provides, subject to certain exceptions, that as long as a shareholder in a public company holds more than 90% of the company’s shares or of a class of shares, that shareholder shall be precluded from purchasing any additional shares unless tendering an offer to purchase all of the outstanding shares of the company or the applicable class of the shares. If the shareholders who do not respond to or accept the offer hold less than 5% of the issued and outstanding share capital of the company or of the applicable class of the shares, and more than half of the shareholders who do not have a personal interest in the offer accept the offer, all of the shares that the acquirer offered to purchase will be transferred to the acquirer by operation of law. However, a tender offer will be accepted if the shareholders who do not accept it hold less than 2% of the issued and outstanding share capital of the company or of the applicable class of the shares.
Upon a successful completion of such a full tender offer, any shareholder that was an offeree in such tender offer, whether such shareholder accepted the tender offer or not, has the right, within six months from the date of acceptance of the tender offer, to petition the court to determine that the tender offer was for less than fair value and that the fair value should be paid as determined by the court. However, under certain conditions, the purchaser may provide in its offer that an offeree who accepted the tender offer will not be entitled to such rights.
If the conditions set forth above are not met, the purchaser may not acquire additional shares of the company from shareholders who accepted the tender offer to the extent that following such acquisition, the purchaser would own more than 90% of the company’s issued and outstanding share capital.
Anti-Takeover Measures under Israeli Law
The Companies Law allows us to create and issue shares having rights different from those attached to our ordinary shares, including shares providing certain preferred rights, distributions or other matters and shares having preemptive rights. As of the date hereof, no preferred shares are authorized under our Articles. In the future, if we do authorize, create and issue a specific class of preferred shares, such class of shares, depending on the specific rights that may be attached to it, may have the ability to frustrate or prevent a takeover or otherwise prevent our shareholders from realizing a potential premium over the market value of their ordinary shares. The authorization and designation of a class of preferred shares will require an amendment to our Articles, which requires the affirmative vote of at least 75% of the voting rights of the Company represented personally or by proxy and voting thereon at a general meeting at which a quorum is present. The convening of the general meeting, the shareholders entitled to participate and the majority vote required to be obtained at such a meeting will be subject to the requirements set forth in the Articles and the Companies Law as described above in “— Shareholder Meetings.”
| 140 |
In addition, certain provisions of the Articles may have the effect of rendering more difficult or discouraging an acquisition of the Company deemed undesirable by the Board. The classification of the Board into three classes with terms of approximately three years each, may make it more difficult for shareholders who oppose the policies of the Board to remove a majority of the then current directors from office quickly. It may also, in some circumstances, together with the other provisions of the Articles and Israeli law, deter or delay potential future merger, acquisition, tender or takeover offers, proxy contests or changes in control or management of the Company.
Changes in Capital
The registered share capital of the Company is NIS 500,000 divided into 50,000,000 ordinary shares, NIS 0.01 par value per share.
Our Articles enable us to increase or reduce our share capital. Any such changes are subject to the provisions of the Companies Law and must be approved by a resolution duly passed by our shareholders at a general meeting by voting on such change in the capital. In addition, transactions that have the effect of reducing capital, such as the declaration and payment of dividends in the absence of sufficient retained earnings or profits and an issuance of shares for less than their nominal value (under certain circumstances), require the approval of both our Board and an Israeli court.
Changes in Shareholder Rights
Pursuant to our Articles, if at any time the share capital is divided into different classes of shares, the Company may by shareholder resolution, unless otherwise provided by the terms of issue of the shares of that class, modify, convert, broaden, add or otherwise alter the rights, privileges, advantages, restrictions and provisions related or unrelated at that time to the shares of any class with the sanction of a resolution passed by a simple majority of those present, personally or by proxy, and voting thereon at a separate general meeting of the holders of the shares of that class. Such majority approval is consistent with Israeli law.
C. Material Contracts
For a description of our material agreements relating to our strategic collaborations and research arrangements and other material agreements, please refer to “Item 4.B. Information on the Company—Business Overview—Strategic Collaborations, Research Arrangements and other Material Agreements.”
Employment Agreements
See “Item 6. Directors, Senior Management and Employees—B. Compensation”.
D. Exchange Controls.
There are no Israeli government laws, decrees, regulations or other legislation that restrict or that affect our export or import of capital, including the availability of cash and cash equivalents for use by us and our wholly-owned subsidiaries, or the remittance of dividends, interest or other payments to non-resident holders of our securities, except for ownership by nationals of certain countries that are, or have been, declared as enemies of Israel or otherwise as set forth under “Item 10. Additional Information—E. Taxation.”
E. Taxation.
The following description is not intended to constitute a complete analysis of all tax consequences relating to the ownership or disposition of our ordinary shares. You should consult your own tax advisor concerning the tax consequences of your particular situation, as well as any tax consequences that may arise under the laws of any state, local, foreign, including Israel, or other taxing jurisdiction.
| 141 |
Certain Israeli Tax Considerations
The following is a brief summary of the material Israeli income tax laws applicable to us. This section also contains a discussion of material Israeli tax consequences concerning the ownership and disposition of our ordinary shares. This summary does not discuss all the aspects of Israeli tax law that may be relevant to a particular investor in light of his or her personal investment circumstances or to some types of investors subject to special treatment under Israeli law. Examples of this kind of investor include residents of Israel or investors in securities who are subject to special tax regimes not covered in this discussion. To the extent that the discussion is based on new tax legislation that has not yet been subject to judicial or administrative interpretation, we cannot assure you that the appropriate tax authorities or the courts will accept the views expressed in this discussion. This summary is based on laws and regulations in effect as of the date hereof and does not take into account possible future amendments which may be under consideration.
General Corporate Tax Structure in Israel
Israeli resident companies (as defined below), such as the Company, are generally subject to corporate tax at the rate of 23% on their taxable income, as of January 1, 2021 (23% in 2020). However, the effective tax rate payable by a company that derives income from a Preferred Enterprise or a Technology Enterprise, as discussed below, may be considerably less.
Capital gains derived by an Israeli resident company are generally subject to tax at the same rate as the corporate tax rate. Under Israeli tax legislation, a corporation will be considered an “Israeli resident” if it meets one of the following: (i) it was incorporated in Israel; or (ii) the control and management of its business are exercised in Israel.
Law for the Encouragement of Industry (Taxes), 5729-1969
The Law for the Encouragement of Industry (Taxes), 5729-1969, which we refer to as the Industry Encouragement Law, provides several tax benefits for “Industrial Companies,” which are defined as Israeli resident-companies which were incorporated in Israel, of which 90% or more of their income in any tax year, other than income from certain government loans, is derived from an “Industrial Enterprise” that it owns and located in Israel or in the “Area”, in accordance with the definition under Section 3A of the Israeli Tax Ordinance. An “Industrial Enterprise” is defined as an enterprise whose principal activity in a given tax year is industrial production. Eligibility for benefits under the Industry Encouragement Law is not contingent upon approval of any governmental authority.
The following tax benefits, among others, are available to Industrial Companies:
| ● | amortization over an eight year period of the cost of purchasing a patent, rights to use a patent and rights to know-how, which are used for the development or advancement of the company, commencing in the year in which such rights were first exercised; | |
| ● | under limited conditions, an election to file consolidated tax returns with related Industrial Companies controlled by it; and | |
| ● | deductions of expenses related to a public offering in equal amounts over a three year period commencing on the year of the offering. |
| 142 |
We believe that we qualify as an “Industrial Company” within the meaning of the Industry Encouragement Law. There can be no assurance that we will continue to qualify as an Industrial Company in the future or that the benefits described above will be available to us at all.
Law for the Encouragement of Capital Investments, 5719-1959
The Law for the Encouragement of Capital Investments, 5719-1959, which we refer to as the Investment Law, provides certain incentives for capital investments in production facilities (or other eligible assets) by “Industrial Enterprises” (as defined under the Investment Law). Generally, an investment program that is implemented in accordance with the provisions of the Investment Law, is entitled to benefits. These benefits may include cash grants from the Israeli government and tax benefits, based upon, among other things, the geographic location in Israel of the facility in which the investment is made. In order to qualify for these incentives, an Approved Enterprise, a Beneficiary Enterprise or a Preferred Enterprise is required to comply with the requirements of the Investment Law.
The Investment Law was significantly amended effective April 1, 2005, further amended as of January 1, 2011, or the 2011 Amendment, and as of January 1, 2017, or the 2017 Amendment. The 2011 Amendment introduced new benefits to replace those granted in accordance with the provisions of the Investment Law in effect prior to the 2011 Amendment. However, companies entitled to benefits under the Investment Law as in effect up to January 1, 2011 were entitled to choose to continue to enjoy such benefits, provided that certain conditions are met, or elect instead, irrevocably, to forego such benefits and elect the benefits of the 2011 Amendment. The 2017 Amendment introduces new benefits for Technological Enterprises, alongside the existing tax benefits.
The following discussion is a summary of the Investment Law following its most recent amendments:
Tax Benefits Under the 2011 Amendment
The 2011 Amendment canceled the availability of the benefits granted to Industrial Companies under the Investment Law prior to 2011 and, instead, introduced new benefits for income generated by a “Preferred Company” through its “Preferred Enterprise” (as such terms are defined in the Investment Law) as of January 1, 2011.
The definition of a Preferred Company includes a company incorporated in Israel that is not fully owned by a governmental entity, and that has, among other things, a Preferred Enterprise and is controlled and managed from Israel. Pursuant to the 2011 Amendment, beginning in 2014 and in each year thereafter until 2016, a Preferred Company may only be entitled to a reduced corporate tax rate of 16% with respect to its preferred income derived by its Preferred Enterprise, unless the Preferred Enterprise is located in a specified development zone, in which case the rate will be 9%. Pursuant to the 2017 Amendment, in 2017 and thereafter, the corporate tax rate for Preferred Enterprise which is located in a specified development zone was reduced to 7.5%, while the reduced corporate tax rate for other development zones remains 16%. Income derived by a Preferred Company from a “Special Preferred Enterprise” (as such term is defined in the Investment Law) would be entitled, during a benefit period of ten years, to further reduced tax rates of 8%, or 5% if the Special Preferred Enterprise is located in a certain development zone. As of January 1, 2017, the definition for ‘Special Preferred Enterprise’ includes less stringent conditions.
As of January 1, 2014, dividends paid to Israeli shareholders out of income attributed to a Preferred Enterprise or to a Special Preferred Enterprise are generally subject to withholding tax at source at the rate of 20% (in the case of non-Israeli shareholders - subject to the receipt in advance of a valid certificate from the ITA allowing for a reduced tax rate, 20%, or such a lower tax rate as may be provided under an applicable tax treaty). However, if such dividends are paid to an Israeli company, no tax is required to be withheld (although, if such dividends are subsequently distributed to individuals or a non-Israeli company, the aforesaid will apply).
| 143 |
New Tax benefits under the 2017 Amendment
The 2017 Amendment was enacted as part of the Economic Efficiency Law that was published on December 29, 2016, and is effective as of January 1, 2017. The 2017 Amendment provides new tax benefits for two types of “Technology Enterprises”, as described below, and is in addition to the other existing tax beneficial programs under the Investment Law.
The 2017 Amendment provides that a technology company satisfying certain conditions will qualify as a “Preferred Technology Enterprise” and will thereby enjoy a reduced corporate tax rate of 12% on income that qualifies as “Preferred Technology Income”, as defined in the Investment Law. The tax rate is further reduced to 7.5% for a Preferred Technology Enterprise located in development zone A. In addition, a Preferred Technology Company will enjoy a reduced corporate tax rate of 12% on capital gain derived from the sale of certain “Benefitted Intangible Assets” (as defined in the Investment Law) to a related foreign company if the Benefitted Intangible Assets were acquired from a foreign company on or after January 1, 2017 for at least NIS 200 million (approximately $56 million), and the sale receives prior approval from the National Authority for Technological Innovation (previously known as the Israeli Office of the Chief Scientist), to which we refer as IIA.
The 2017 Amendment further provides that a technology company satisfying certain conditions will qualify as a “Special Preferred Technology Enterprise” and will thereby enjoy a reduced corporate tax rate of 6% on “Preferred Technology Income” regardless of the company’s geographic location within Israel. In addition, a Special Preferred Technology Enterprise will enjoy a reduced corporate tax rate of 6% on capital gain derived from the sale of certain “Benefitted Intangible Assets” to a related foreign company if the Benefitted Intangible Assets were either developed by the Special Preferred Technology Enterprise or acquired from a foreign company on or after January 1, 2017, and the sale received prior approval from IIA. A Special Preferred Technology Enterprise that acquires Benefitted Intangible Assets from a foreign company for more than NIS 500 million will be eligible for these benefits for at least ten years, subject to certain approvals as specified in the Investment Law.
Dividends distributed by a Preferred Technology Enterprise or a Special Preferred Technology Enterprise to Israeli shareholders, paid out of Preferred Technology Income, are subject to withholding tax at source at the rate of 20% (in the case of non-Israeli shareholders - subject to the receipt in advance of a valid certificate from the ITA allowing for a reduced tax rate, 20%, or such lower rate as may be provided in an applicable tax treaty). However, if such dividends are paid to an Israeli company, no tax is required to be withheld (although, if such dividends are subsequently distributed to individuals or a non-Israeli company, the aforesaid will apply). If such dividends are distributed to a foreign parent company holding, alone or together with other foreign companies, at least 90% of the shares of the distributing company and other conditions are met, the withholding tax rate will be 4% (or a lower rate under a tax treaty, if applicable, subject to the receipt in advance of a valid certificate from the ITA allowing for a reduced tax rate).
After examining the impact of the 2017 Amendment, we submitted a request to receive a tax ruling from the Israel Tax Authority to be recognized as a Preferred Technology Enterprise and we received a tax ruling from the Israel Tax Authority granting GRD a Preferred Technology Enterprise status, subject to terms and conditions determined in the tax ruling.
Taxation of Our Israeli Individual Shareholders on Receipt of Dividends
Israeli residents who are individuals are generally subject to Israeli income tax for dividends paid on our ordinary shares (other than bonus shares or share dividends) at a rate of 25%, or 30% if the recipient of such dividend is a Substantial Shareholder (as defined below) at the time of distribution or at any time during the preceding 12-month period. However, dividends distributed from taxable income accrued from Preferred Enterprise or Preferred Technology Enterprise to Israeli individuals are subject to withholding tax at the rate of 20%. However, if such dividends are distributed to an Israeli company, no tax is imposed (although, if such dividends are subsequently distributed to individuals or a non-Israeli company, withholding tax at a rate of 20% or such lower rate as may be provided in an applicable tax treaty (subject to the receipt in advance of a valid certificate from the Israel Tax Authority (“ITA”) allowing for a reduced tax rate will apply). An average rate will be set in case the dividend is distributed from mixed types of income (regular and preferred income).
| 144 |
A “Substantial Shareholder” is generally a person who alone, or together with his or her relative or another person who collaborates with him or her on a regular basis, holds, directly or indirectly, at least 10% of any of the “means of control” of a corporation. “Means of control” generally include the right to vote, receive profits, nominate a director or an officer, receive assets upon liquidation or instruct someone who holds any of the aforesaid rights regarding the manner in which he or she is to exercise such right(s), all regardless of the source of such right.
With respect to individuals, the term “Israeli resident” is generally defined under Israeli tax legislation as a person whose center of life is in Israel. The Israeli Tax Ordinance (as amended by Amendment Law No. 132 of 2002), states that in order to determine the center of life of an individual, consideration will be given to the individual’s family, economic and social connections, including: (i) place of permanent residence; (ii) place of residential dwelling of the individual and the individual’s immediate family; (iii) place of the individual’s regular or permanent occupation or the place of his or her permanent employment; (iv) place of the individual’s active and substantial economic interests; (v) place of the individual’s activities in organizations, associations and other institutions. The center of life of an individual will be presumed to be in Israel if: (i) the individual was present in Israel for 183 days or more in the tax year; or (ii) the individual was present in Israel for 30 days or more in the tax year, and the total period of the individual’s presence in Israel in that tax year and the two previous tax years is 425 days or more. Such presumption may be rebutted either by the individual or by the assessing officer.
Payers of dividends on our ordinary shares, including the Israeli stockbroker effectuating the transaction, or the financial institution through which the securities are held, are generally required, subject to any of the foregoing exemptions, reduced tax rates and the demonstration of a shareholder regarding his, her or its foreign residency, to withhold tax upon the distribution of dividend at the rate of 25% (whether the recipient is a Substantial Shareholder or not), so long as the shares are registered with a nominee company.
Taxation of Israeli Resident Corporations on Payment of Dividends
Israeli resident corporations are generally exempt from Israeli corporate income tax with respect to dividends paid on ordinary shares of Israeli resident corporations as long as the profits out of which the dividends were paid were derived in Israel.
Capital Gains Taxes Applicable to Israeli Resident Shareholders
The income tax rate applicable to real capital gains derived by an Israeli individual resident from the sale of shares that were purchased after January 1, 2012, whether listed on a stock exchange or not, is 25%. However, if such shareholder is considered a Substantial Shareholder at the time of sale or at any time during the preceding 12 month period and/or claims a deduction for interest and linkage differences expenses in connection with the purchase and holding of such shares, such gain will be taxed at the rate of 30%.
Moreover, capital gains derived by an individual shareholder who is a dealer or trader in securities, or to whom such income is otherwise taxable as ordinary business income, are taxed in Israel at their marginal rates applicable to business income (up to 50% in 2020 and 2021, including Excess Tax as detailed below).
At the sale of securities traded on a stock exchange, a detailed return, including a computation of the tax due, must be filed and an advanced payment must be paid on January 31 and July 31 of every tax year in respect of sales of securities made within the previous six months. However, if all tax due was withheld at source according to applicable provisions of the Israeli Tax Ordinance and regulations promulgated thereunder, the aforementioned return is not required to be filed and no advance payment must be paid. Capital gain is also reportable on the annual income tax return.
| 145 |
Taxation of Non-Israeli Shareholders on Receipt of Dividends
Non-Israeli residents are generally subject to Israeli income tax on the receipt of dividends paid on our ordinary shares at the rate of 25% (or 30% for individuals, if such person is a Substantial Shareholder at the time he or she receives the dividend or on any date in the 12 months preceding such date), or 20% if the dividend is distributed from income attributed to Preferred Enterprise unless a lower rate is provided under an applicable tax treaty between Israel and the shareholder’s country of residence and provided that a certificate from the Israel Tax Authority allowing for a reduced withholding tax rate is obtained in advance.
A non-Israeli resident who has dividend income derived from or accrued in Israel, from which the full amount of tax was withheld at source, is generally exempt from the duty to file tax returns in Israel in respect of such income; provided that (i) such income was not derived from a business conducted in Israel by the taxpayer, (ii) the taxpayer has no other taxable sources of income in Israel with respect to which a tax return is required to be filed, and (iii) the taxpayer is not obligated to pay excess tax (as further explained below).
For example, under the Convention Between the Government of the United States of America and the Government of Israel with Respect to Taxes on Income, as amended, or the U.S.-Israel Tax Treaty, Israeli withholding tax on dividends paid to a U.S. resident for treaty purposes may not, in general, exceed 25%, subject to certain conditions. Where the recipient is a U.S. corporation owning 10% or more of the voting shares of the paying corporation during the part of the paying corporation’s taxable year which precedes the date of payment of the dividend and during the entirety of its prior taxable year (if any), the Israeli tax withheld may not exceed 12.5%, subject to certain conditions.
Payers of dividends on our ordinary shares, including the Israeli stockbroker effectuating the transaction, or the financial institution through which the securities are held, are generally required, subject to any of the foregoing exemptions, reduced tax rates and the demonstration of a shareholder regarding his, her or its foreign residency, to withhold tax upon the distribution of dividend at the rate of 25% (whether the recipient is a Substantial Shareholder or not), so long as the shares are registered with a nominee company.
Capital Gains Income Taxes Applicable to Non-Israeli Shareholders
Non-Israeli resident shareholders are generally exempt from Israeli capital gains tax on any gains derived from the sale, exchange or disposition of our ordinary shares, provided that such shareholders did not acquire their shares prior to January 1, 2009 or acquired their shares after the Company was listed for trading on NASDAQ and such gains were not derived from a permanent business or business activity of such shareholders in Israel. These provisions dealing with capital gain are not applicable to a person whose gains from selling or otherwise disposing of the shares are deemed to be business income. However, non-Israeli corporations will not be entitled to the foregoing exemptions if an Israeli resident (i) has a controlling interest of more than 25% in such non-Israeli corporation or (ii) is the beneficiary of or is entitled to 25% or more of the revenues or profits of such non-Israeli corporation, whether directly or indirectly.
In addition, a sale of securities by a non-Israeli resident may be exempt from Israeli capital gains tax under the provisions of an applicable tax treaty. For example, under the U.S.-Israel Tax Treaty, the sale, exchange or disposition of our ordinary shares by a shareholder who is a U.S. resident (for purposes of the U.S.-Israel Tax Treaty) holding the ordinary shares as a capital asset and is entitled to claim the benefits afforded to such a resident by the U.S.-Israel Tax Treaty, or a Treaty U.S. Resident, is generally exempt from Israeli capital gains tax unless: (i) such Treaty U.S. Resident is an individual and was present in Israel for 183 days or more in the aggregate during the relevant taxable year; (ii) such Treaty U.S. Resident holds, directly or indirectly, shares representing 10% or more of our voting power of the Company during any part of the 12 month period preceding such sale, exchange or disposition, subject to certain conditions; (iii) the capital gains arising from such sale, exchange or disposition are attributable to a permanent establishment of the Treaty U.S. Resident maintained in Israel, subject to certain conditions; (iv) the capital gains arising from such sale, exchange or disposition is attributed to real estate located in Israel; or (v) the capital gains arising from such sale, exchange or disposition is attributed to royalties. In any such case, the sale, exchange or disposition of our ordinary shares would be subject to Israeli tax, to the extent applicable. However, under the U.S.-Israel Tax Treaty, such Treaty U.S. Resident would be permitted to claim a credit for such taxes against U.S. federal income tax imposed on any gain from such sale, exchange or disposition, under the circumstances and subject to the limitations specified in the U.S.-Israel Income Tax Treaty.
| 146 |
Regardless of whether shareholders may be liable for Israeli income tax on the sale of our ordinary shares, the payment of the consideration may be subject to withholding of Israeli tax at the source. Accordingly, shareholders may be required to demonstrate that they are exempt from tax on their capital gains in order to avoid withholding at source at the time of sale. Specifically, in transactions involving a sale of all of the shares of an Israeli resident company, in the form of a merger or otherwise, the Israel Tax Authority may require from shareholders who are not liable for Israeli tax to sign declarations in forms specified by this authority or obtain a specific exemption from the Israel Tax Authority to confirm their status as non-Israeli resident, and, in the absence of such declarations or exemptions, may require the purchaser of the shares to withhold taxes at source.
Excess Tax
Individuals who are subject to tax in Israel are also subject to an additional tax at a rate of 3% on annual income exceeding a certain threshold (NIS 647,640 for 2021, which amount is linked to the annual change in the Israeli consumer price index), including, but not limited to, dividends, interest and capital gains.
Estate and Gift Tax
Israeli law presently does not impose estate or gift taxes.
Certain U.S. Federal Income Tax Considerations
The following is a general summary of certain material U.S. federal income tax consequences relating to the purchase, ownership and disposition of our ordinary shares by U.S. Holders (as defined below). This summary is based on the Code, the regulations of the U.S. Department of the Treasury issued pursuant to the Code, or the Treasury Regulations, the income tax treaty between the United States and Israel, or the U.S.-Israel Tax Treaty, and administrative and judicial interpretations thereof, all as in effect on the date hereof and all of which are subject to change, possibly with retroactive effect, or to different interpretation. No ruling has been sought from the Internal Revenue Service, or the IRS, with respect to any U.S. federal income tax consequences described below, and there can be no assurance that the IRS or a court will not take a contrary position. This summary is no substitute for consultation by prospective investors with their own tax advisors and does not constitute tax advice. This summary applies only to U.S. Holders that hold our ordinary shares as capital assets for U.S. federal income tax purposes (generally, property held for investment) and does not address all of the tax considerations that may be relevant to specific U.S. Holders in light of their particular circumstances or to U.S. Holders subject to special treatment under U.S. federal income tax law (including, without limitation, banks, insurance companies, tax-exempt entities, retirement plans, regulated investment companies, partnerships, dealers in securities, brokers, real estate investment trusts, certain former citizens or residents of the United States, persons who acquire our ordinary shares as part of a straddle, hedge, conversion transaction or other integrated investment, persons who acquire our ordinary shares through the exercise or cancellation of employee stock options or otherwise as compensation for their services, persons that have a “functional currency” other than the U.S. dollar, persons that own (or are deemed to own, indirectly, or by attribution) 10% or more of our shares (by vote or value), or persons that mark their securities to market for U.S. federal income tax purposes). This summary does not address any U.S. state or local or non-U.S. tax considerations, any U.S. federal estate, gift or alternative minimum tax considerations, or any U.S. federal tax consequences other than U.S. federal income tax consequences.
| 147 |
As used in this summary, the term “U.S. Holder” means a beneficial owner of our ordinary shares that is, for U.S. federal income tax purposes, (i) an individual citizen or resident of the United States, (ii) a corporation, or other entity taxable as a corporation for U.S. federal income tax purposes, created or organized in or under the laws of the United States, any state thereof, or the District of Columbia, (iii) an estate the income of which is subject to U.S. federal income tax regardless of its source, or (iv) a trust with respect to which a court within the United States is able to exercise primary supervision over its administration and one or more U.S. persons have the authority to control all of its substantial decisions, or that has a valid election in effect under applicable Treasury Regulations to be treated as a “United States person.”
If an entity or arrangement treated as a partnership for U.S. federal income tax purposes holds our ordinary shares, the tax treatment of such entity or arrangement treated as a partnership and each person treated as a partner thereof generally will depend upon the status and activities of the entity and such person. A holder that is treated as a partnership for U.S. federal income tax purposes should consult its own tax advisor regarding the U.S. federal income tax considerations applicable to it and its partners of the purchase, ownership and disposition of our ordinary shares.
Prospective investors should be aware that this summary does not address the tax consequences to investors who are not U.S. Holders. Prospective investors should consult their own tax advisors as to the particular tax considerations applicable to them relating to the purchase, ownership and disposition of our ordinary shares, including the applicability of U.S. federal, state and local tax laws and non-U.S. tax laws.
Taxation of U.S. Holders
Distributions. Subject to the discussion below under “Passive Foreign Investment Company,” a U.S. Holder that receives a distribution with respect to an ordinary share generally will be required to include the amount of such distribution in gross income as a dividend (without reduction for any Israeli tax withheld from such distribution) when actually or constructively received to the extent of the U.S. Holder’s pro rata share of our current and/or accumulated earnings and profits (as determined under U.S. federal income tax principles). Any distributions in excess of our earnings and profits will be applied against and will reduce (but not below zero) the U.S. Holder’s tax basis in its ordinary shares, and, to the extent they exceed that tax basis, will be treated as gain from the sale or exchange of our ordinary shares. We do not intend to calculate our earnings and profits under U.S. federal income tax principles. Therefore, a U.S. Holder should expect that a distribution will be treated as a dividend even if that distribution would otherwise be treated as a non-taxable return of capital or as capital gain under the rules described above.
As noted above, we do not anticipate paying any cash dividends in the foreseeable future. If we were to pay dividends, we expect to pay such dividends in NIS. A dividend paid in NIS, including the amount of any Israeli taxes withheld, will be includible in a U.S. Holder’s income at a U.S. dollar amount calculated by reference to the exchange rate in effect on the date such dividend is received, regardless of whether the payment is in fact converted into U.S. dollars. If the dividend is converted to U.S. dollars on the date of receipt, a U.S. Holder generally will not recognize a foreign currency gain or loss. However, if the U.S. Holder converts the NIS into U.S. dollars on a later date, the U.S. Holder must include, in computing its income, any gain or loss resulting from any exchange rate fluctuations. The gain or loss will be equal to the difference between (i) the U.S. dollar value of the amount included in income when the dividend was received and (ii) the amount received on the conversion of the NIS into U.S. dollars. Such gain or loss generally will be ordinary income or loss and will be U.S. source income or loss for U.S. foreign tax credit purposes. U.S. Holders should consult their own tax advisors regarding the tax consequences to them if we pay dividends in NIS or any other non-U.S. currency.
| 148 |
Subject to certain significant conditions and limitations, any Israeli taxes paid on or withheld from distributions from us and not refundable to a U.S. Holder may be credited against the U.S. Holder’s U.S. federal income tax liability or, alternatively, may be deducted from the U.S. Holder’s taxable income. The election to deduct, rather than credit, foreign taxes, is made on a year-by-year basis and applies to all foreign taxes paid by a U.S. Holder or withheld from a U.S. Holder that year. Dividends paid on our ordinary shares generally will constitute income from sources outside the United States and be categorized as “passive category income” or, in the case of some U.S. Holders, as “general category income” for U.S. foreign tax credit purposes. Because the rules governing foreign tax credits are complex, U.S. Holders should consult their own tax advisors regarding the availability of foreign tax credits in their particular circumstances.
Dividends paid on our ordinary shares will not be eligible for the “dividends-received” deduction generally allowed to corporate U.S. Holders with respect to dividends received from U.S. corporations.
Certain distributions treated as dividends that are received by an individual U.S. Holder from a “qualified foreign corporation” may be classified as “qualified dividend income,” — which is generally taxed at the lower applicable long term capital gains rates provided certain holding period and other requirements are satisfied. A non-U.S. corporation (other than a PFIC for the taxable year in which the dividend is paid or the preceding taxable year) generally will be considered to be a qualified foreign corporation (i) if it is eligible for the benefits of a comprehensive tax treaty with the United States which the Secretary of Treasury of the United States determines is satisfactory for purposes of this provision and which includes an exchange of information program, or (ii) with respect to any dividend it pays on stock which is readily tradable on an established securities market in the United States. As discussed below under “Passive Foreign Investment Company,” we believe that we were a PFIC for our 2021 taxable year and expect to be a PFIC for the 2022 taxable year. Because the PFIC determination is highly fact intensive, there can be no assurance that we will be a PFIC in 2022 or for any other taxable year. Our ordinary shares will generally be considered to be readily tradable on an established securities market in the United States if they are listed on the Nasdaq Capital Market, as we intend our ordinary shares will be. U.S. Holders should consult their own tax advisors regarding the availability of the lower rate for dividends paid with respect to our ordinary shares.
The additional 3.8% “net investment income tax” (described below) may apply to dividends received by certain U.S. Holders who meet certain modified adjusted gross income thresholds.
Sale, Exchange or Other Taxable Disposition of Ordinary Shares. Subject to the discussion under “Passive Foreign Investment Company” below, a U.S. Holder generally will recognize capital gain or loss upon the sale, exchange, or other taxable disposition of our ordinary shares in an amount equal to the difference between the amount realized on the sale, exchange, or other taxable disposition and the U.S. Holder’s adjusted tax basis (determined under U.S. federal income tax rules) in such ordinary shares. This capital gain or loss will be long-term capital gain or loss if the U.S. Holder’s holding period in our ordinary shares exceeds one year. Preferential tax rates for long-term capital gain (currently, with a maximum rate of 20%) will apply to individual U.S. Holders. The deductibility of capital losses is subject to limitations. The gain or loss generally will be income or loss from sources within the United States for U.S. foreign tax credit purposes, subject to certain possible exceptions under the U.S.-Israel Tax Treaty. The additional 3.8% “net investment income tax” (described below) may apply to gains recognized upon the sale, exchange, or other taxable disposition of our ordinary shares by certain U.S. Holders who meet certain modified adjusted gross income thresholds.
U.S. Holders should consult their own tax advisors regarding the U.S. federal income tax consequences of receiving currency other than U.S. dollars upon the disposition of their ordinary shares.
Passive Foreign Investment Company. In general, a non-U.S. corporation will be treated as a PFIC for U.S. federal income tax purposes in any taxable year in which either (i) at least 75% of its gross income is “passive income,” or (ii) on average at least 50% of its assets by value produce passive income or are held for the production of passive income. Passive income for this purpose generally includes, among other things, certain dividends, interest, royalties, rents and gains from commodities and securities transactions and from the sale or exchange of property that gives rise to passive income. Passive income also includes amounts derived by reason of the temporary investment of funds, including those raised in a public offering. Assets that produce or are held for the production of passive income may include cash, even if held as working capital or raised in a public offering, as well as marketable debt securities and other assets that may produce passive income. In determining whether a non-U.S. corporation is a PFIC, a proportionate share of the income and assets of each corporation in which it owns, directly or indirectly, at least a 25% interest (by value) is taken into account.
| 149 |
A foreign corporation’s PFIC status is an annual determination that is based on tests that are factual in nature, and our status for any year will depend on our income, assets, and activities for such year. Based upon our review of our financial data, we believe that we were a PFIC for our 2021 taxable year and expect to be a PFIC for the 2022 taxable year. Because PFIC status is determined annually and is based on our income, assets and activities for the entire taxable year, it is not possible to determine with certainty whether we will be characterized as a PFIC for the 2022 taxable year until after the close of the year, and there can be no assurance that we will not be classified as a PFIC in any future year.
Default PFIC Rules. If we are a PFIC for any tax year, a U.S. Holder who does not make a timely “qualified electing fund” election, or “QEF election” or a mark-to-market election (as described below), referred to in this summary as a “Non-Electing U.S. Holder,” will be subject to special rules with respect to (i) any “excess distribution” (generally, the portion of any distributions received by the Non-Electing U.S. Holder on the ordinary shares in a taxable year in excess of 125% of the average annual distributions received by the Non-Electing U.S. Holder in the three preceding taxable years, or, if shorter, the Non-Electing U.S. Holder’s holding period for the ordinary shares), and (ii) any gain realized on the sale or other disposition of such ordinary shares. Under these rules:
| ● | the excess distribution or gain would be allocated ratably over the Non-Electing U.S. Holder’s holding period for such ordinary shares; | |
| ● | the amount allocated to the current taxable year and any year prior to us becoming a PFIC would be taxed as ordinary income; and | |
| ● | the amount allocated to each of the other taxable years would be subject to tax at the highest rate of tax in effect for the applicable class of taxpayer for that year, and an interest charge for the deemed deferral benefit would be imposed with respect to the resulting tax attributable to each such other taxable year. |
If a Non-Electing U.S. Holder who is an individual dies while owning our ordinary shares, the Non-Electing U.S. Holder’s successor would be ineligible to receive a step-up in tax basis of such ordinary shares. Non-Electing U.S. Holders should consult their tax advisors regarding the application of the “net investment income tax” (described below) to their specific situation.
To the extent a distribution on our ordinary shares does not constitute an excess distribution to a Non-Electing U.S. Holder, such Non-Electing U.S. Holder generally will be required to include the amount of such distribution in gross income as a dividend to the extent of our current and/or accumulated earnings and profits (as determined for U.S. federal income tax purposes) that are not allocated to excess distributions. The tax consequences of such distributions are discussed above under “Taxation of U.S. Holders—Distributions.” Each U.S. Holder is encouraged to consult its own tax advisor with respect to the appropriate U.S. federal income tax treatment of any distribution on our ordinary shares.
If we are treated as a PFIC for any taxable year during the holding period of a Non-Electing U.S. Holder, we will continue to be treated as a PFIC for all succeeding years during which the Non-Electing U.S. Holder is treated as a direct or indirect Non-Electing U.S. Holder even if we are not a PFIC for such years. A U.S. Holder is encouraged to consult its tax advisor with respect to any available elections that may be applicable in such a situation, including the “deemed sale” election of Section 1298(b)(1) of the Code (which will be taxed under the adverse tax rules described above).
| 150 |
We may invest in the equity of foreign corporations that are PFICs or may own subsidiaries that own PFICs. If we are classified as a PFIC, under attribution rules, U.S. Holders will be subject to the PFIC rules with respect to their indirect ownership interests in such PFICs, such that a disposition of the ordinary shares of the PFIC or receipt by us of a distribution from the PFIC generally will be treated as a deemed disposition of such ordinary shares or the deemed receipt of such distribution by the U.S. Holder, subject to taxation under the PFIC rules. There can be no assurance that a U.S. Holder will be able to make a QEF election or a mark-to-market election with respect to PFICs in which we invest. Each U.S. Holder is encouraged to consult its own tax advisor with respect to tax consequences of an investment by us in a corporation that is a PFIC.
QEF Election. Certain adverse consequences of PFIC status can be mitigated for holders of our ordinary shares if a U.S. Holder makes a QEF election. A U.S. Holder who makes a timely QEF election, referred to in this disclosure as an “Electing U.S. Holder,” with respect to us must report for U.S. federal income tax purposes its pro rata share of our ordinary earnings and net capital gain, if any, for our taxable year that ends with or within the taxable year of the Electing U.S. Holder. The “net capital gain” of a PFIC is the excess, if any, of the PFIC’s net long-term capital gains over its net short-term capital losses. The amount so included in income generally will be treated as ordinary income to the extent of such Electing U.S. Holder’s allocable share of the PFIC’s ordinary earnings and as long-term capital gain to the extent of such Electing U.S. Holder’s allocable share of the PFIC’s net capital gains. Such Electing U.S. Holder generally will be required to translate such income into U.S. dollars based on the average exchange rate for the PFIC’s taxable year with respect to the PFIC’s functional currency. Such income generally will be treated as income from sources outside the United States for U.S. foreign tax credit purposes. Amounts previously included in income by such Electing U.S. Holder under the QEF rules generally will not be subject to tax when they are distributed to such Electing U.S. Holder. The Electing U.S. Holder’s tax basis in our ordinary shares generally will increase by any amounts so included under the QEF rules and decrease by any amounts not included in income when distributed.
An Electing U.S. Holder will be subject to U.S. federal income tax on such amounts for each taxable year in which we are a PFIC, regardless of whether such amounts are actually distributed to such Electing U.S. Holder. However, an Electing U.S. Holder may, subject to certain limitations, elect to defer payment of current U.S. federal income tax on such amounts, subject to an interest charge. If an Electing U.S. Holder is an individual, any such interest will be treated as non-deductible “personal interest.”
Any net operating losses or net capital losses of a PFIC will not pass through to the Electing U.S. Holder and will not offset any ordinary earnings or net capital gain of a PFIC recognized by Electing U.S. Holder in subsequent years.
So long as an Electing U.S. Holder’s QEF election with respect to us is in effect with respect to the entire holding period for our ordinary shares, any gain or loss recognized by such Electing U.S. Holder on the sale, exchange or other disposition of such shares generally will be long-term capital gain or loss if such Electing U.S. Holder has held such shares for more than one year at the time of such sale, exchange or other disposition. Preferential tax rates for long-term capital gain (currently, a maximum rate of 20%) will apply to individual U.S. Holders. The deductibility of capital losses is subject to limitations.
In general, a U.S. Holder must make a QEF election on or before the due date for filing its income tax return for the first year to which the QEF election is to apply. A U.S. Holder makes a QEF election by completing the relevant portions of and filing IRS Form 8621 in accordance with the instructions thereto. Upon request, we expect to provide U.S. Holders with the information needed to complete IRS Form 8621 (which form would be required to be filed with the IRS on an annual basis by the U.S. Holder) and to make and maintain a valid QEF election for any year in which we or any of our subsidiaries that we control is a PFIC. There is no assurance, however, that we will have timely knowledge of our status as a PFIC, or that the information that we provide will be adequate to allow U.S. Holders to make a QEF election. A QEF election will not apply to any taxable year during which we are not a PFIC, but will remain in effect with respect to any subsequent taxable year in which we become a PFIC.
| 151 |
Each U.S. Holder should consult its own tax advisor with respect to the advisability of, the tax consequences of, and the procedures for making a QEF election with respect to us.
Mark-to-Market Election. Alternatively, if our ordinary shares are treated as “marketable stock,” a U.S. Holder would be allowed to make a “mark-to-market” election with respect to our ordinary shares, provided the U.S. Holder completes and files IRS Form 8621 in accordance with the relevant instructions and related Treasury Regulations. If that election is made, the U.S. Holder generally would include as ordinary income in each taxable year the excess, if any, of the fair market value of our ordinary shares at the end of the taxable year over such holder’s adjusted tax basis in such ordinary shares. The U.S. Holder would also be permitted an ordinary loss in respect of the excess, if any, of the U.S. Holder’s adjusted tax basis in our ordinary shares over their fair market value at the end of the taxable year, but only to the extent of the net amount previously included in income as a result of the mark-to- market election. A U.S. Holder’s tax basis in our ordinary shares would be adjusted to reflect any such income or loss amount. Gain realized on the sale, exchange or other disposition of our ordinary shares would be treated as ordinary income, and any loss realized on the sale, exchange or other disposition of our ordinary shares would be treated as ordinary loss to the extent that such loss does not exceed the net mark-to-market gains previously included in income by the U.S. Holder, and any loss in excess of such amount will be treated as capital loss. Amounts treated as ordinary income will not be eligible for the favorable tax rates applicable to qualified dividend income or long-term capital gains.
Generally, stock will be considered marketable stock if it is “regularly traded” on a “qualified exchange” within the meaning of applicable Treasury Regulations. A class of stock is regularly traded on an exchange during any calendar year during which such class of stock is traded, other than in de minimis quantities, on at least 15 days during each calendar quarter. To be marketable stock, our ordinary shares must be regularly traded on a qualifying exchange (i) in the United States that is registered with the SEC or a national market system established pursuant to the Exchange Act or (ii) outside the United States that is properly regulated and meets certain trading, listing, financial disclosure and other requirements. Our ordinary shares are expected to constitute “marketable stock” as long as they remain listed on the Nasdaq Capital Market and are regularly traded.
A mark-to-market election will not apply to our ordinary shares held by a U.S. Holder for any taxable year during which we are not a PFIC, but will remain in effect with respect to any subsequent taxable year in which we become a PFIC. Such election will not apply to any PFIC subsidiary that we own. Each U.S. Holder is encouraged to consult its own tax advisor with respect to the availability and tax consequences of a mark-to-market election with respect to our ordinary shares.
Each U.S. Holder should consult its own tax adviser with respect to the applicability of the “net investment income tax” (discussed below) where a mark-to-market election is in effect.
In addition, U.S. Holders should consult their tax advisors regarding the IRS information reporting and filing obligations that may arise as a result of the ownership of ordinary shares in a PFIC, including IRS Form 8621, Information Return by a Shareholder of a Passive Foreign Investment Company or Qualified Electing Fund.
The U.S. federal income tax rules relating to PFICs, QEF elections, and mark-to market elections are complex. U.S. Holders are urged to consult their own tax advisors with respect to the purchase, ownership and disposition of our ordinary shares, any elections available with respect to such ordinary shares and the IRS information reporting obligations with respect to the purchase, ownership and disposition of our ordinary shares.
| 152 |
Certain Reporting Requirements
Certain U.S. Holders may be required to file IRS Form 926, Return by U.S. Transferor of Property to a Foreign Corporation and IRS Form 5471, Information Return of U.S. Persons With Respect to Certain Foreign Corporations, reporting transfers of cash or other property to us and information relating to the U.S. Holder and us. Substantial penalties may be imposed upon a U.S. Holder that fails to comply. See also the discussion regarding Form 8621, Information Return by a Shareholder of a Passive Foreign Investment Company or Qualified Electing Fund, above.
In addition, certain U.S. Holders must report information on IRS Form 8938, Statement of Specified Foreign Financial Assets, with respect to their investments in certain “specified foreign financial assets,” which would include an investment in our ordinary shares, if the aggregate value of all of those assets exceeds $50,000 on the last day of the taxable year (and in some circumstances, a higher threshold). This reporting requirement applies to individuals and certain U.S. entities.
U.S. Holders who fail to report required information could become subject to substantial penalties. U.S. Holders should consult their tax advisors regarding the possible implications of these reporting requirements arising from their investment in our ordinary shares.
Backup Withholding Tax and Information Reporting Requirements
Generally, information reporting requirements will apply to distributions on our ordinary shares or proceeds on the disposition of our ordinary shares paid within the United States (and, in certain cases, outside the United States) to U.S. Holders other than certain exempt recipients, such as corporations. Furthermore, backup withholding (currently at 24%) may apply to such amounts if the U.S. Holder fails to (i) provide a correct taxpayer identification number, (ii) report interest and dividends required to be shown on its U.S. federal income tax return, or (iii) make other appropriate certifications in the required manner. U.S. Holders who are required to establish their exempt status generally must provide such certification on IRS Form W-9.
Backup withholding is not an additional tax. Amounts withheld as backup withholding from a payment may be credited against a U.S. Holder’s U.S. federal income tax liability and such U.S. Holder may obtain a refund of any excess amounts withheld by filing the appropriate claim for refund with the IRS and furnishing any required information in a timely manner.
Medicare Tax on Investment Income
Certain U.S. persons, including individuals, estates and trusts, will be subject to an additional 3.8% Medicare tax, or “net investment income tax,” on unearned income. For individuals, the additional net investment income tax applies to the lesser of (i) “net investment income” or (ii) the excess of “modified adjusted gross income” over $200,000 ($250,000 if married and filing jointly or $125,000 if married and filing separately). “Net investment income” generally equals the taxpayer’s gross investment income reduced by the deductions that are allocable to such income. Investment income generally includes, among other things, passive income such as interest, dividends, annuities, royalties, rents, and capital gains. U.S. Holders are urged to consult their own tax advisors regarding the implications of the additional net investment income tax resulting from their ownership and disposition of our ordinary shares.
THE DISCUSSION ABOVE IS A GENERAL SUMMARY. IT DOES NOT COVER ALL TAX MATTERS THAT MAY BE OF IMPORTANCE TO A PROSPECTIVE INVESTOR. EACH PROSPECTIVE INVESTOR IS URGED TO CONSULT ITS OWN TAX ADVISOR ABOUT THE TAX CONSEQUENCES RELATING TO THE PURCHASE, OWNERSHIP AND DISPOSITION OF OUR ORDINARY SHARES IN LIGHT OF THE INVESTOR’S OWN CIRCUMSTANCES, INCLUDING THE CONSEQUENCES OF ANY PROPOSED CHANGE IN APPLICABLE LAWS.
| 153 |
F. Dividends and Paying Agents.
Not applicable.
G. Statements by Experts.
Not applicable.
H. Documents on Display.
The SEC maintains an Internet website that contains reports and other information regarding issuers that file electronically with the SEC. You may read and copy this annual report, including the related exhibits and schedules, and any document we file with the SEC at http://www.sec.gov.
As a “foreign private issuer,” we are subject to the information reporting requirements of the Exchange Act that are applicable to foreign private issuers, and under those requirements file reports with the SEC. Those other reports or other information may be inspected without charge at the locations described above. As a “foreign private issuer,” we are exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders will be exempt from the reporting and “short-swing” profit recovery provisions contained in Section 16 of the Exchange Act with respect to their purchases and sales of ordinary shares. Furthermore, as a “foreign private issuer,” we are also not subject to the requirements of Regulation FD (Fair Disclosure) promulgated under the Exchange Act.
We maintain a corporate website at http://www.galmedpharma.com. Information contained on, or that can be accessed through, our website is not incorporated by reference into this annual report and does not constitute a part of this annual report. We have included our website address in this annual report solely as an inactive textual reference.
I. Subsidiary Information.
Not applicable.
ITEM 11. Quantitative and Qualitative Disclosures About Market Risk.
Quantitative and Qualitative Disclosure About Market Risk
We are exposed to market risks in the ordinary course of our business. Market risk represents the risk of loss that may impact our financial position, results of operations or cash flows due to adverse changes in financial market prices and rates, including interest rates and foreign exchange rates, of financial instruments.
Foreign Currency Exchange Risk
Our foreign currency exposures give rise to market risk associated with exchange rate movements of the Euro and NIS mainly against the U.S. dollar because a large portion of our expenses are denominated in Euros and NIS. Our Euro expenses consist principally of payments made to sub-contractors and consultants for pre-clinical studies, clinical trials and other research and development activities. Our NIS expenses consist principally of payments made to employees, subcontractors and consultants for pre-clinical studies, clinical trials, professional services, other research and development activities and general and administrative activities. We anticipate that a large portion of our expenses will continue to be denominated in currencies other than the U.S. dollar. Our financial position, results of operations and cash flow are subject to fluctuations due to changes in foreign currency exchange rates. Our results of operations and cash flow are, therefore, subject to fluctuations due to changes in foreign currency exchange rates and may be adversely affected in the future due to changes in foreign exchange rates. Approximately 25% of our expected expenses are denominated in NIS. Changes of 5% and 10% in the U.S. dollar to NIS exchange rate will increase/decrease our operation expenses by 1.25% and 2.5%, respectively. Approximately 10% of our expected expenses are denominated in Euros, and another 10% are denominated in GBP. Changes of 5% and 10% in the U.S. dollar to Euro exchange rate and in the U.S. dollar to GBP exchange rate, will increase/decrease our operation expenses by 1.0% and 2.0%, respectively. To date, fluctuations in the exchange rates have not materially affected our results of operations or financial condition for the periods under review.
| 154 |
To date, we have not engaged in hedging our foreign currency exchange risk. In the future, we may enter into formal currency hedging transactions to decrease the risk of financial exposure from fluctuations in the exchange rates of our principal operating currencies. These measures, however, may not adequately protect us from the material adverse effects of such fluctuations.
Interest Rate Risk
Our primary exposure to market risk is interest income sensitivity, which is affected by changes in the general level of U.S. interest rates. We currently do not hedge interest rate exposure. Because of the short-term maturities of our cash equivalents and investment securities, we do not believe that an increase in market rates would have any significant impact on the realized value of our investment securities. If a 10% change in interest rates were to have occurred on December 31, 2021, this change would not have had a material effect on the fair value of our investment portfolio as of that date.
Liquidity
We do not believe that our cash and cash equivalents and available for sale investments have significant risk of default or illiquidity. While we believe our cash, cash equivalents and available for sale investments do not contain excessive risk, we cannot provide absolute assurance that in the future our investments will not be subject to adverse changes in market value. In addition, we maintain significant amounts of cash and cash equivalents at one or more financial institutions that are in excess of federally insured limits.
ITEM 12. Description of Securities Other Than Equity Securities.
A. Debt Securities.
Not applicable.
B. Warrants and Rights.
Not applicable.
C. Other Securities.
Not applicable.
D. American Depositary Shares.
Not applicable.
| 155 |
PART II
ITEM 13. Defaults, Dividend Arrearages and Delinquencies.
Not applicable.
ITEM 14. Material Modifications to the Rights of Security Holders and Use of Proceeds.
Not applicable.
ITEM 15. Controls and Procedures.
Disclosure Controls and Procedures
We performed an evaluation of the effectiveness of our disclosure controls and procedures that are designed to ensure that information required to be disclosed in this annual report and filed with the SEC is recorded, processed, summarized and reported timely within the time period specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by an issuer in the reports that it files or submits under the Exchange Act, is accumulated and communicated to the issuer’s management, including its principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure. There can be no assurance that our disclosure controls and procedures will detect or uncover all failures of persons within our Company to disclose information otherwise required to be set forth in our reports. Nevertheless, our disclosure controls and procedures are designed to provide reasonable assurance of achieving the desired control objectives. Based on our evaluation, our management, including our President, Chief Executive Officer and Chairman and Chief Accounting Officer, have concluded that our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15(d)-15(e) of the Exchange Act) as of the end of the period covered by this annual report are effective at such reasonable assurance level.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over our financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated under the Exchange Act as a process designed by, or under the supervision of, the company’s principal executive and principal financial officers and effected by the company’s board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
| ● | pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transaction and dispositions of the assets of the company; | |
| ● | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and | |
| ● | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’s assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
| 156 |
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2021. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework (2013). Based on that assessment, our management concluded that as of December 31, 2021, our internal control over financial reporting was effective.
Attestation Report of the Registered Public Accounting Firm
The effectiveness of our internal control over financial reporting as of December 31, 2021 has been audited by Brightman Almagor Zohar & Co., a Firm in the Deloitte Global Network, an independent registered public accounting firm, as stated in their report included elsewhere in this annual report.
Changes in Internal Controls Over Financial Reporting
There were no changes in our internal control over financial reporting that occurred during the year ended December 31, 2021 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
ITEM 16. [RESERVED]
ITEM 16A. Audit Committee Financial Expert.
Our Board has determined that Mr. Poshinski qualifies as an audit committee financial expert pursuant to the applicable SEC rules and that Mr. Poshinski is “independent” in accordance with the Nasdaq Capital Market corporate governance requirements. For information relating to Mr. Poshinski’s qualifications and experience, see “Item 6. Directors, Senior Management and Employees—A. Directors and Senior Management.”
ITEM 16B. Code of Ethics.
We have adopted a Code of Business Conduct and Ethics applicable to all of our directors and employees, including our President, Chief Executive Officer and Chairman, Chief Financial Officer, controller or principal accounting officer or other persons performing similar functions, which is a “code of ethics” as defined in Item 16B of Form 20-F promulgated by the SEC and as required by the Nasdaq Listing Rules, which refers to Section 406(c) of the Sarbanes-Oxley Act. Section 406(c) of the Sarbanes-Oxley Act provides that a “code of ethics” means such standards as are reasonably necessary to promote (i) honest and ethical conduct, including the ethical handling of actual or apparent conflicts of interest between personal and professional relationships; (ii) full, fair, accurate, timely and understandable disclosure in the periodic reports required to be filed by the issuer; and (iii) compliance with applicable governmental rules and regulation.
The full text of the Code of Business Conduct and Ethics is posted on our website at www.galmedpharma.com. Information contained on, or that can be accessed through, our website does not constitute a part of this prospectus and is not incorporated by reference herein. We will provide a copy of such code of ethics without charge upon request by mail or by telephone. If we make any amendment to the Code of Business Conduct and Ethics or grant any waivers, including any implicit waiver, from a provision of the Code of Business Conduct and Ethics, we will disclose the nature of such amendment or waiver on our website to the extent required by the rules and regulations of the SEC.
| 157 |
ITEM 16C. Principal Accountant Fees and Services.
Brightman Almagor Zohar & Co., a Firm in the Deloitte Global Network, an independent registered public accounting firm, served as our independent public accountants for the fiscal years ended December 31, 2021 and 2020, for which audited financial statements appear in this annual report.
The following table presents the aggregate fees for professional services rendered by such accountants to us during their respective term as our principal accountants in 2021 and 2020.
| 2021 | 2020 | |||||||
| (US$ in thousands) | (US$ in thousands) | |||||||
| Audit Fees (1) | 120 | 100 | ||||||
| Audit-Related fees (2) | 20 | 20 | ||||||
| Tax Fees (3) | 16 | 12 | ||||||
| Total | 156 | 132 | ||||||
| (1) | Includes professional services rendered in connection with the audit of our annual financial statements and the review of our interim financial statements. |
| (2) | Audit related services consist of services that were reasonably related to the performance of the audit or reviews of our financial statements and not included under “Audit Fees” above, including, principally, providing consents for registration statement filings. |
| (3) | Tax fees consist of services related to obtaining a tax ruling and applying for a grant. |
Audit Committee Pre-Approval Policies and Procedures
One of our audit committee’s main roles is to assist the board of directors in fulfilling its responsibility for oversight of the quality and integrity of the accounting, auditing and reporting practices of the Company. The audit committee oversees the appointment, compensation, and oversight of the public accounting firm engaged to prepare or issue an audit report on the financial statements of the Company. Our Board has delegated to the audit committee the power to pre-approve non-auditing services rendered by the Company’s independent auditors without the need for further approval by the board of directors. As such, our audit committee has adopted a pre-approval policy for the engagement of our independent registered public accounting firm to perform certain audit and non-audit services. Pursuant to this policy, which is designed to assure that such engagements do not impair the independence of our auditors, the audit committee pre-approves annually a list of specific audit and non-audit services in the categories of audit services, audit-related services, tax services and other services that may be performed by our independent registered public accounting firm. The last pre-approval policy was adopted by our audit committee on April 24, 2022 for a period of twelve months. Since its establishment in May 2014, the audit committee has approved all of the audit-related fees, tax fees and all other fees. If a type of service that is to be provided by our auditors has not received such general pre-approval, it will require specific pre-approval by our audit committee. The policy prohibits retention of the independent registered public accounting firm to perform the prohibited non-audit functions defined in applicable SEC rules.
| 158 |
ITEM 16D. Exemptions from the Listing Standards for Audit Committees.
Not applicable.
ITEM 16E. Purchases of Equity Securities by the Issuer and Affiliated Purchasers.
Not applicable.
ITEM 16F. Change in Registrant’s Certifying Accountant.
Not applicable.
ITEM 16G. Corporate Governance.
Our shares are listed on the Nasdaq Capital Market under the symbol “GLMD.” In addition to the corporate governance requirements of the Sarbanes-Oxley Act and the related rules implemented by the SEC, we must comply with the Nasdaq Listing Rules. Under those Nasdaq Listing Rules, we may elect to follow certain corporate governance practices permitted under the Companies Law in lieu of compliance with corresponding corporate governance requirements otherwise imposed by the Nasdaq Listing Rules for U.S. domestic issuers.
In accordance with Israeli law and practice, and subject to the exemption set forth in Rule 5615 of the Nasdaq Listing Rules, we follow the provisions of the Companies Law, rather than the Nasdaq Listing Rules, with respect to the following requirements:
| ● | Distribution of certain reports to shareholders. As opposed to the Nasdaq Listing Rules, which require listed issuers to make certain reports, such as annual reports, interim reports and quarterly reports, available to shareholders in one of a number of specific manners, Israeli law does not require us to distribute periodic reports directly to shareholders, and the generally accepted business practice in Israel is not to distribute such reports to shareholders, but to make such reports available through a public website. In addition to making such reports available on a public website, we plan to make our audited financial statements available to our shareholders at our offices and will only mail such reports to shareholders upon request. As a foreign private issuer, we are generally exempt from the SEC’s proxy solicitation rules. See “Item 10. Additional Information—Documents on Display” for a description of our Exchange Act reporting obligations. | |
| ● | Quorum. While the Nasdaq Listing Rules require that the quorum for purposes of any meeting of the holders of a listed company’s common voting stock be no less than 33.33% of the company’s outstanding common voting stock, under Israeli law, a company is entitled to determine in its articles of association the number of shareholders and percentage of holdings required for a quorum at a shareholders meeting. Our articles of association provide that a quorum of two or more shareholders holding at least 33.33% of the voting rights in person or by proxy is required for commencement of business at a general meeting. However, the quorum set forth in our articles of association with respect to an adjourned meeting consists of any two shareholders present in person or by proxy even if, between them, they represent shares conferring 33.33% or less of the voting rights of the Company. | |
| ● | Nomination of directors. With the exception of directors elected by our Board due to vacancy, our directors are elected by an annual meeting of our shareholders to hold office until the next annual meeting following three years from his or her election. See “Item 6. Directors, Senior Management and Employees—C. Board Practices.” The nominations for directors, which are presented to our shareholders by our Board, are made by the nominating committee itself, in accordance with the provisions of Nasdaq Capital Market Listing Rule 5605(e), our Articles and the Companies Law. |
| 159 |
| ● | Compensation of officers. We follow the provisions of the Companies Law with respect to matters in connection with the composition and responsibilities of our remuneration committee, Office Holder compensation and any required approval by the shareholders of such compensation. Israeli law and our Articles do not require that the independent members of our Board, or a remuneration committee composed solely of independent members of our Board, determine an executive officer’s compensation, as is generally required under the Nasdaq Listing Rules with respect to the Chief Executive Officer and all other executive officers of a company. Instead, remuneration of Office Holders is determined and approved by our remuneration committee, and in general, by our Board as well, and in certain circumstances, by our shareholders, as detailed above. The requirements for shareholder approval of any Office Holder compensation, and the relevant majority or Special Majority for such approval, are all as set forth in the Companies Law. Thus, we seek shareholder approval for all corporate actions with respect to Office Holder compensation requiring such approval under the requirements of the Companies Law, including for our Compensation Policy and for certain Office Holder Compensation, rather than seeking approval for such corporate actions in accordance with Nasdaq Listing Rules. All members of our remuneration committee are independent directors under applicable Nasdaq Capital Market and SEC rules, as affirmatively determined by our Board. See “Item 6. Directors, Senior Management and Employees—B. Compensation.” | |
| ● | Independent directors. Although Israeli law does not require that a majority of the directors serving on our Board be “independent,” as defined under Nasdaq Capital Market Listing Rule 5605(a)(2), but rather requires we have at least two external directors who meet the requirements of the Companies Law, as described above under “Item 6. Directors, Senior Management and Employees—C. Board Practices—External Directors.”, following our “opt-out” of the requirement to appoint external directors, a majority of our Board is independent based on the Nasdaq Capital Market rules. We are required, however, to ensure that all members of our audit committee are “independent” under the applicable Nasdaq Capital Market and SEC criteria for independence (as we cannot exempt ourselves from compliance with that SEC independence requirement, despite our status as a foreign private issuer). Our independent directors’ conduct regularly scheduled meetings at which only such independent directors are present, as required by the Nasdaq Listing Rules. Our Board has affirmatively determined that each of Mr. Nir, Mr. Poshinski, Dr. Sidransky and Dr. Brosgart qualifies as “independent” under the Nasdaq Capital Market independence standards. | |
| ● | Shareholder approval. We will seek shareholder approval for all corporate actions requiring such approval under requirements of the Companies Law, rather than seeking approval for corporate actions in accordance with Nasdaq Capital Market Listing Rule 5635. In particular, under this Nasdaq Capital Market rule, shareholder approval is generally required for: (i) an acquisition of shares or assets of another company that involves the issuance of 20% or more of the acquirer’s shares or voting rights or if a director, officer or 5% shareholder has greater than a 5% interest in the target company or the consideration to be received; (ii) the issuance of shares leading to a change of control; (iii) adoption or amendment of equity compensation arrangements; and (iv) issuances of 20% or more of the shares or voting rights (including securities convertible into, or exercisable for, equity) of a listed company via a private placement (or via sales by directors, officers or 5% shareholders) if such equity is issued (or sold) at below the greater of the book or market value of shares. By contrast, under the Companies Law, shareholder approval is required for, among other things: (i) transactions with directors concerning the terms of their service or indemnification, exemption and insurance for their service (or for any other position that they may hold at a company), for which approvals of the remuneration committee, board of directors and shareholders are all required, (ii) Extraordinary Transactions with controlling shareholders of publicly held companies, which require the special approval described under “Item 6. Directors, Senior Management and Employees—C. Board Practices—Approval of Related Party Transactions under Israeli Law—Transactions with Controlling Shareholders,” and (iii) terms of office and employment or other engagement of the controlling shareholder of the Company or such controlling shareholder’s relative, which require the special approval described under “Item 6. Directors, Senior Management and Employees—B. Compensation” and “Item 6. Directors, Senior Management and Employees—C. Board Practices—Approval of Related Party Transactions under Israeli Law.” In addition, under the Companies Law, a merger requires approval of the shareholders of each of the merging companies. See also “Compensation of officers” above. |
| 160 |
ITEM 16H. Mine Safety Disclosure.
Not applicable.
ITEM 16I. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
Not applicable.
PART III
ITEM 17. Financial Statements.
We have responded to Item 18 in lieu of responding to this item.
ITEM 18. Financial Statements.
Please refer to the financial statements beginning on page F-1. The following financial statements, financial statement schedules and related notes are filed as part of this annual report, together with the report of the independent registered public accounting firm.
| 161 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the shareholders and the Board of Directors of Galmed Pharmaceuticals Ltd.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Galmed Pharmaceuticals Ltd. and subsidiaries (the “Company”) as of December 31, 2021 and 2020, the related consolidated statements of operations, comprehensive loss, shareholders’ equity and cash flows, for each of the three years in the period ended December 31, 2021, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2021 and 2020, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2021, in conformity with accounting principles generally accepted in the United States of America.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of December 31, 2021, based on criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated May 2, 2022, expressed an unqualified opinion on the Company’s internal control over financial reporting.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current-period audit of the financial statements that was communicated or required to be communicated to the audit committee and that (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Valuation of Marketable Debt Securities Classified as Available-for-Sale– Refer to Note 3 to the consolidated financial statements
Critical Audit Matter Description
The Company invests its excess cash primarily in available-for-sale marketable debt securities. Investments in marketable debt securities classified as available-for-sale are reported at fair value in the financial statements. The investments totaled $31.9 million at December 31, 2021.
| F-1 |
We identified the valuation of investments in marketable debt securities classified as available-for-sale as a critical audit matter because of the magnitude of these investments and due to the increased extent of audit effort in relation to our audit as a whole.
How the Critical Audit Matter Was Addressed in the Audit
Our audit procedures related to the valuation of the fair value of investments in marketable debt securities classified as available-for-sale included the following, among others:
| ● | We tested the effectiveness of the Company’s controls over the valuation of investments in marketable debt securities classified as available-for-sale, including an assessment of the relevant controls at each service organization over the determination of fair value. |
| ● | With the assistance of our specialists experienced in the valuation of securities, we obtained independent estimates of the fair value of the investments in marketable debt securities classified as available-for-sale held by the Company as of December 31, 2021 and compared our estimates to the Company’s estimates. |
| ● | We agreed the recorded values of investments in marketable debt securities classified as available-for-sale to the fair values indicated in the service organizations’ balance confirmations. |
/s/
Brightman Almagor Zohar & Co.
Certified Public Accountants
A Firm in the Deloitte Global Network
May 2, 2022
We have served as the Company’s auditor since 2013.
| F-2 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the shareholders and the Board of Directors of Galmed Pharmaceuticals Ltd.
Opinion on Internal Control over Financial Reporting
We have audited the internal control over financial reporting of Galmed Pharmaceuticals Ltd. and subsidiaries (the “Company”) as of December 31, 2021, based on criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2021, based on criteria established in Internal Control — Integrated Framework (2013) issued by COSO.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated financial statements as of and for the year ended December 31, 2021, of the Company and our report dated May 2, 2022, expressed an unqualified opinion on those consolidated financial statements.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control over Financing Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of the consolidated financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of the consolidated financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the consolidated financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Brightman Almagor Zohar & Co.
Brightman Almagor Zohar & Co.
Certified Public Accountants
A Firm in the Deloitte Global Network
Tel Aviv, Israel
May 2, 2022
| F-3 |
GALMED PHARMACEUTICALS LTD.
Consolidated Balance Sheets
U.S. Dollars in thousands, except share data and per share data
| As of December 31, | |||||||||||
| 2021 | 2020 | ||||||||||
| Assets | |||||||||||
| Current assets | |||||||||||
| Cash and cash equivalents | $ | $ | |||||||||
| Restricted cash | 8 | ||||||||||
| Short-term deposits | - | ||||||||||
| Marketable debt securities | 3 | ||||||||||
| Other accounts receivable | 4 | ||||||||||
| Total current assets | |||||||||||
| Operating lease right-of-use assets | 5 | ||||||||||
| Property and equipment, net | 6 | ||||||||||
| Total non-current assets | |||||||||||
| Total assets | $ | $ | |||||||||
| Liabilities and stockholders’ equity | |||||||||||
| Current liabilities | |||||||||||
| Trade payables | $ | $ | |||||||||
| Other accounts payable | |||||||||||
| Total current liabilities | |||||||||||
| Non-current liabilities | |||||||||||
| Operating lease liabilities, net of current portion | 5 | $ | $ | ||||||||
| Total non-current liabilities | |||||||||||
| Stockholders’ equity | |||||||||||
| Ordinary shares, par value NIS per share; Authorized shares; Issued and outstanding: shares as of December 31, 2021; | 9 | ||||||||||
| Additional paid-in capital | |||||||||||
| Accumulated other comprehensive income (loss) | ( | ) | |||||||||
| Accumulated deficit | ( | ) | ( | ) | |||||||
| Total stockholders’ equity | |||||||||||
| Total liabilities and stockholders’ equity | $ | $ | |||||||||
Accompanying notes are an integral part of the consolidated financial statements.
| F-4 |
GALMED PHARMACEUTICALS LTD.
Consolidated Statements of Operations
U.S. Dollars in thousands, except share data and per share data
| Year ended December 31, | |||||||||||||||
| 2021 | 2020 | 2019 | |||||||||||||
| Research and development expenses | 10 | ||||||||||||||
| General and administrative expenses | 11 | ||||||||||||||
| Total operating loss | |||||||||||||||
| Financial income, net | 12 | ( | ) | ( | ) | ( | ) | ||||||||
| Net loss | $ | $ | $ | ||||||||||||
| Basic and diluted net loss per share | $ | $ | $ | ||||||||||||
| Weighted-average number of shares outstanding used in computing basic and diluted net loss per share | |||||||||||||||
Accompanying notes are an integral part of the consolidated financial statements.
| F-5 |
GALMED PHARMACEUTICALS LTD.
Consolidated Statements of Comprehensive Loss
U.S. Dollars in thousands, except share data and per share data
| Year ended December 31, | ||||||||||||
| 2021 | 2020 | 2019 | ||||||||||
| Net loss | $ | $ | $ | |||||||||
| Other comprehensive loss: | ||||||||||||
| Net unrealized loss (gain) on available for sale securities | ( | ) | ( | ) | ||||||||
| Comprehensive loss | $ | $ | $ | |||||||||
Accompanying notes are an integral part of the consolidated financial statements.
| F-6 |
GALMED PHARMACEUTICALS LTD.
Statements of Changes in Stockholders’ Equity
U.S. Dollars in thousands, except share data and per share data
| Accumulated | ||||||||||||||||||||||||
| Additional | other | |||||||||||||||||||||||
| Ordinary shares | paid-in | comprehensive | Accumulated | |||||||||||||||||||||
| Shares | Amount | capital | income (loss) | deficit | Total | |||||||||||||||||||
| Balance - January 1, 2020 | $ | $ | $ | $ | ( | ) | $ | |||||||||||||||||
| Stock-based compensation | — | |||||||||||||||||||||||
| Exercise of options and vesting of restricted stock units | (*) | |||||||||||||||||||||||
| Issuance
of ordinary shares in at-the-market (“ATM”) offering, net of $ | (*) | |||||||||||||||||||||||
| Unrealized gain from marketable debt securities | — | |||||||||||||||||||||||
| Net loss | — | ( | ) | ( | ) | |||||||||||||||||||
| Balance - December 31, 2020 | $ | $ | $ | $ | ( | ) | $ | |||||||||||||||||
| Stock-based compensation | — | |||||||||||||||||||||||
| Exercise of options and vesting of restricted stock units | (*) | (*) | (*) | |||||||||||||||||||||
| Issuance
of ordinary shares in ATM offering, net of $ | ||||||||||||||||||||||||
| Issuance
of ordinary shares under Underwritten Public Offering agreement, net of $ | ||||||||||||||||||||||||
| Unrealized loss on marketable debt securities | — | ( | ) | ( | ) | |||||||||||||||||||
| Net loss | — | ( | ) | ( | ) | |||||||||||||||||||
| Balance - December 31, 2021 | $ | $ | $ | ( | ) | $ | ( | ) | $ | |||||||||||||||
| (*) | |
| (**) |
Accompanying notes are an integral part of the consolidated financial statements.
| F-7 |
GALMED PHARMACEUTICALS LTD.
Consolidated Statements of Cash Flows
U.S. Dollars in thousands, except share data and per share data
| Year ended December 31, | ||||||||||||
| 2021 | 2020 | 2019 | ||||||||||
| Cash flow from operating activities | ||||||||||||
| Net loss for the year | $ | ( | ) | $ | ( | ) | $ | ( | ) | |||
| Adjustments required to reconcile net loss to net cash used in operating activities: | ||||||||||||
| Depreciation and amortization | ||||||||||||
| Amortization of discount (premium) on marketable debt securities | ( | ) | ||||||||||
| Gain on sale of marketable debt securities | ( | ) | ( | ) | ( | ) | ||||||
| Finance expenses | ||||||||||||
| Interest income from short-term deposits | ( | ) | ( | ) | ||||||||
| Stock-based compensation expense | ||||||||||||
| Changes in operating assets and liabilities: | ||||||||||||
| Decrease (increase) in other accounts receivable | ( | ) | ( | ) | ||||||||
| Increase (decrease) in trade payables | ( | ) | ||||||||||
| Increase (decrease) in other accounts payable | ( | ) | ||||||||||
| Net cash used in operating activities | ( | ) | ( | ) | ( | ) | ||||||
| Cash flow from investing activities | ||||||||||||
| Purchase of property and equipment | ( | ) | ( | ) | ( | ) | ||||||
| Increase in restricted deposit | ( | ) | - | - | ||||||||
| Investment in securities, available for sale | ( | ) | ( | ) | ( | ) | ||||||
| Proceeds from sale of securities, available for sale | ||||||||||||
| Proceeds from (investment in) short-term deposits, net | ( | ) | ||||||||||
| Net cash provided by investing activities | ||||||||||||
| Cash flow from financing activities | ||||||||||||
| Issuance of ordinary shares in at-the-market offering, net of issuance costs (**) | - | - | ||||||||||
| Issuance of ordinary shares, net of issuance costs (**) | - | |||||||||||
| Proceeds from exercise of options | (*) | |||||||||||
| Net cash provided by financing activities | ||||||||||||
| Decrease in cash, cash equivalents and restricted cash | ( | ) | ( | ) | ( | ) | ||||||
| Cash and cash equivalents and restricted cash at the beginning of the year | ||||||||||||
| Cash, cash equivalents and restricted cash at the end of the year | $ | $ | $ | |||||||||
| Supplemental disclosure of cash flow information: | ||||||||||||
| Cash received from interest | $ | $ | $ | |||||||||
| Non-cash transactions: | ||||||||||||
| Recognition of right-of-use asset and lease liabilities from adoption of ASU 2016-02, net | $ | $ | $ | |||||||||
| Right-of-use assets obtained in exchange for new operating lease liabilities, net | $ | $ | $ | |||||||||
| (*) | |
| (**) |
The accompanying notes are an integral part of the consolidated financial statements.
| F-8 |
GALMED PHARMACEUTICALS LTD.
Notes to Consolidated Financial Statements
Note 1 – General
Galmed
Pharmaceuticals Ltd. (the “Company”) was incorporated in Israel on
The Company holds a wholly-owned subsidiary, Galmed International Ltd., which was incorporated in Malta. Galmed International Ltd. previously held a wholly-owned subsidiary, Galmed Medical Research Ltd., which was incorporated in Israel, and had been an inactive company since 2015 and was liquidated in February 2019.
The Company also holds two additional wholly-owned subsidiaries, Galmed Research and Development Ltd and Galtopa Therapeutics Ltd., both of which are incorporated in Israel.
The
Company is a clinical-stage biopharmaceutical company primarily focused on the development of therapeutics for the treatment of liver
diseases. The Company has an operating history limited to pre-clinical and clinical drug development. To date, the Company has focused
almost exclusively on developing its product candidates, Aramchol and Amilo-5MER. The Company funded its research and development programs
and operations to date primarily through proceeds from private placements and public offerings. The Company currently has no products
approved for marketing and has not generated any revenue from product sales to date. As of December 31, 2021, the Company had cash and
cash equivalents of $
The
Company has incurred operating losses in each year since inception. The Company’s loss attributable to holders of its ordinary
shares for the years ended December 31, 2019, 2020, and 2021 was approximately $
The Company will need to raise substantial, additional capital to fund its operations and to develop Aramchol for, and beyond its current development stage and any future commercialization, as well as any additional indications.
Based on the Company’s current operating plan, the Company’s management currently estimates that its cash position will support its current clinical trials and operations as currently conducted for more than 12 months from the date of issuance of these financial statements.
Note 2 – Significant Accounting Policies
| A. | Basis of presentation |
The consolidated financial statements have been prepared in accordance with United States Generally Accepted Accounting Principles (“U.S. GAAP”).
| B. | Use of estimates |
The preparation of consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities as of the date of the financial statements, and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
| F-9 |
GALMED PHARMACEUTICALS LTD.
Notes to Consolidated Financial Statements
Note 2 – Significant Accounting Policies (Cont.)
| C. | Financial statement in U.S. dollars |
The functional currency of the Company and its subsidiaries is in U.S dollar (the “dollar”), because the dollar is the currency of the primary economic environment in which the Company and its subsidiaries operate, and expect to continue operating in the foreseeable future. Transactions and balances denominated in dollars are presented in their original amounts. Non-dollar denominated transactions and balances have been re-measured to dollars in accordance with the provisions of ASC 830-10, “Foreign Currency Translation.” All transaction gains and losses from re-measurement of monetary balance sheet items denominated in non-dollar currencies are reflected in the statement of operations as financial income or expenses, as appropriate.
| D. | Principles of consolidation |
The consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries: Galmed Research and Development Ltd., Galmed International Ltd. and Galtopa Therapeutics Ltd. All intercompany balances and transactions have been eliminated upon consolidation.
| E. | Cash and cash equivalents |
Cash equivalents are short-term, highly liquid investments that are readily convertible into cash with maturities of three months or less as of the date acquired.
| F. | Restricted Cash |
Cash that is held for a specific purpose and is not available for immediate or general business use due to external restrictions is classified in our consolidated balance sheets as restricted cash.
| G. | Marketable debt securities |
The Company invests most of its excess cash primarily in debt securities.
The Company accounts for its investments in investment grade debt securities in accordance with ASC 320 “Investments - Debt and Equity Securities”. Management determines the appropriate classification of its investments in debt securities at the time of purchase and re-evaluates such determinations at each balance sheet date.
Marketable debt securities are considered to be available for sale and are carried at fair value on the consolidated balance sheet. Unrealized gains and losses net of tax, if any, are reported in a separate component of shareholders’ equity in accumulated other comprehensive income (“OCI”). Gains and losses are recognized when realized, on a specific identification basis, in the Company’s consolidated statements of operations.
Following the adoption of ASC 326 in January 2020, current expected credit losses on the Company’s marketable grade debt securities are recorded, if expected, through an allowance for current expected credit losses. The amount of allowance for current expected credit losses is limited to the amount that the fair value is less than the amortized cost basis. Any remaining unrealized losses are included in accumulated other comprehensive loss in shareholders’ equity.
If the Company intends to sell the debt security (that is, it has decided to sell the security), or more likely than not will be required to sell the security before recovery of its amortized cost basis, any allowance for current expected credit losses is written off and the amortized cost basis shall be written down to the debt security’s fair value at the reporting date with any incremental impairment reported in earnings. Based on management’s assessment, the Company does not intend to sell its securities for less than amortized cost; therefore, an allowance for current expected credit losses has not been recorded as of December 31, 2021 and 2020.
| F-10 |
GALMED PHARMACEUTICALS LTD.
Notes to Consolidated Financial Statements
Note 2 – Significant Accounting Policies (Cont.)
| H. | Concentrations of credit risk |
Financial instruments which potentially subject the Company to credit risk consist primarily of cash, cash equivalents, marketable securities and short-term deposits. The Company hold these investments in highly-rated financial institutions, and, by policy, limit the amounts of credit exposure to any one financial institution. These amounts at times may exceed federally insured limits. The Company has not experienced any credit losses in such accounts and do not believe we are exposed to any significant credit risk on these funds. The Company has no off-balance sheet concentrations of credit risk, such as foreign currency exchange contracts, option contracts, or other hedging arrangements.
| I. | Property and equipment |
Property and equipment are stated at cost, less accumulated depreciation. Depreciation is calculated using the straight-line method over the estimated useful lives of the assets. The annual depreciation rates are as follows:
| % | ||||
| Office furniture and equipment | ||||
| Computer software and electronic equipment | ||||
| Leasehold improvements |
| J. | Impairment of long-lived assets |
The Company’s and its subsidiaries’ long-lived assets are reviewed for impairment in accordance with ASC 360-10, “Accounting for the Impairment or Disposal of Long-Lived Assets,” whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of the assets to the future undiscounted cash flows expected to be generated by the assets. If such assets are considered to be impaired, the impairment to be recognized is measured by the amount by which the carrying amount of the assets exceeds their fair value. During 2021 and 2020, no impairment losses were recorded.
| K. | Severance pay |
The
Company employees are included under section 14 of the Severance Compensation Act, 1963 (“Section 14”) in Israel for a portion
of their salaries. According to Section 14, these employees are entitled to monthly deposits at a rate of
| F-11 |
GALMED PHARMACEUTICALS LTD.
Notes to Consolidated Financial Statements
Note 2 – Significant Accounting Policies (Cont.)
| L. | Fair value of financial instruments |
The estimated fair value of financial instruments was determined by the Company using available market information and valuation methodologies. Considerable judgment is required in estimating fair values. Accordingly, the estimates may not be indicative of the amounts the Company could realize in a current market exchange.
The following methods and assumptions were used by the Company in estimating its fair value disclosures for financial instruments:
The carrying amounts of cash and cash equivalents, short-term bank deposits, other accounts receivables, trade payables and other trade payables approximate their fair value due to the short-term maturity of such instruments.
Fair value is an exit price representing the amount that would be received upon selling an asset or that would be paid to transfer a liability in an orderly transaction between market participants. As such, fair value is a market-based measurement that should be determined based on assumptions used by market participants in pricing an asset or a liability.
A three-tier fair-value hierarchy was established as a basis for considering such assumptions and for inputs used in the valuation methodologies in measuring fair value:
| ● | Level 1 - Observable inputs that reflect quoted prices (unadjusted) for identical assets or liabilities in active markets | |
| ● | Level 2 - Other inputs that are directly or indirectly observable in the marketplace; and | |
| ● | Level 3 - Unobservable inputs that are supported by little or no market activity |
The fair value hierarchy also requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value.
The Company’s marketable debt securities are measured at fair value on a recurring basis by level within the fair value hierarchy. Other than the marketable debt securities, which includes corporate bonds and mutual funds as of December 31, 2021, the Company does not have any other financial assets or financial liabilities marked to market at fair value.
The fair value of the Company’s marketable debt securities measured at fair value on a recurring basis by level within the fair value hierarchy are as follows (in thousands):
| December 31, 2021 | ||||||||||||||||
| Fair | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Value | |||||||||||||
| Marketable debt securities | $ | $ | $ | |||||||||||||
| December 31, 2020 | ||||||||||||||||
| Fair | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Value | |||||||||||||
| Marketable debt securities | $ | $ | $ | |||||||||||||
| M. | Accounting for stock-based compensation |
The Company applies ASC 718-10, “Share-Based Payment,” which requires the measurement and recognition of compensation expense for all share-based payment awards made to employees and directors, including employee stock options under the Company’s stock plans, based on estimated fair values. ASC 718-10 requires companies to estimate the fair value of equity-based payment awards on the date of grant using an option-pricing model. The value of the portion of the award that is ultimately expected to vest is recognized as expense over the requisite service periods in the Company’s consolidated statement of operations.
| F-12 |
GALMED PHARMACEUTICALS LTD.
Notes to Consolidated Financial Statements
Note 2 – Significant Accounting Policies (Cont.)
| M. | Accounting for stock-based compensation (Cont.) |
All issuances of stock options or other equity instruments to non-employees as consideration for goods or services received by the Company are accounted for based on the fair value of the equity instruments issued.
The Company estimates the fair value of restricted shares based on the market price of the shares at the grant date and estimates the fair value of stock options granted using a . The option-pricing model requires a number of assumptions, the most significant of which are the expected stock-price volatility and the expected option term (the time from the grant date until the options are exercised or expire).
The Company’s calculations of the expected volatility were based upon actual historical stock-price movements over the period, which was equal to the expected option term. The expected option term was calculated for options granted to employees and directors in accordance with ASC-718-10-S99, using the “simplified” method, and grants to non-employees were based on the contractual term. Historically, the Company has not paid dividends, and has no foreseeable plans to do so. The risk-free interest rate is based on the yield from U.S. Treasury zero-coupon bonds with an equivalent term.
| N. | Revenue Recognition |
The Company only has one license agreement for which it has recognized revenues to date, from a license agreement with Samil Pharm. Co., Ltd. (“Samil” or “Samil Agreement”). The Samil Agreement was signed on July 28, 2016, for an exclusive, royalty-bearing license for the commercialization of Aramchol (with an option to manufacture) for the treatment of fatty liver indications including NASH in the Republic of Korea. Additionally, following the ARREST Study, Samil has an option to extend the License to Vietnam, which, if exercised, would increase the clinical- and regulatory-based milestone payments.
Under
the terms of the Samil Agreement, the Company received an up-front payment of approximately $
In accordance with ASC 606 the Company determined that the Agreement included a combined performance obligation representing the delivery of the exclusive license and completion of the ARREST study.
As of December 31, 2021, management evaluated the remaining clinical and regulatory milestones and determined that the variable consideration should not be recorded as revenue for the period ended December 31, 2021. The Company will re-evaluate the transaction price in each reporting period when events whose outcomes are resolved or other changes in circumstances occur that would indicate it is appropriate to recognize variable consideration as revenue.
| F-13 |
GALMED PHARMACEUTICALS LTD.
Notes to Consolidated Financial Statements
Note 2 – Significant Accounting Policies (Cont.)
| O. | Research and development expenses |
Research and development expenses are charged to the statement of operations as incurred.
| P. | Income taxes |
The Company accounts for income taxes utilizing the asset and liability method in accordance with ASC 740, “Income Taxes.” Current tax liabilities are recognized for the estimated taxes payable on tax returns for the current year. Deferred tax liabilities or assets are recognized for the estimated future tax effects attributable to temporary differences between the income-tax bases of assets and liabilities and their reported amounts in the financial statements and for tax loss carry forwards. Measurement of current and deferred tax liabilities and assets is based on provisions of enacted tax laws, and deferred tax assets are reduced, if necessary, by the amount of tax benefits, the realization of which is not considered more likely than not based on available evidence. As of December 31, 2021, and 2020, the Company had a full valuation allowance against deferred tax assets.
The Company is subject to the provisions of ASC 740-10-25, “Income Taxes” (“ASC 740”). ASC 740 prescribes a more likely-than-not threshold for the financial statement recognition of uncertain tax positions. ASC 740 clarifies the accounting for income taxes by prescribing a minimum recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. On a yearly basis, the Company undergoes a process to evaluate whether income tax accruals are in accordance with ASC 740 guidance on uncertain tax positions. The Company has not recorded any liability for uncertain tax positions for the years ended December 31, 2021 and 2020.
| Q. | Basic and diluted net loss per share |
Basic net loss per share is computed based on the weighted-average number of shares outstanding during each year. Diluted net loss per share is computed based on the weighted-average number of shares outstanding during each year, plus the dilutive potential of the ordinary shares considered outstanding during the year, in accordance with ASC 260-10, “Earnings Per Share.”
All outstanding stock options and warrants were excluded from the calculation of the diluted loss per share for the years ended December 31, 2021, 2020 and 2019, because all such securities have an anti-dilutive effect.
| F-14 |
GALMED PHARMACEUTICALS LTD.
Notes to Consolidated Financial Statements
Note 2 – Significant Accounting Policies (Cont.)
| R. | Segment Reporting |
The chief operating decision maker for the Company is the Chief Executive Officer. The Chief Executive Officer reviews financial information presented on a consolidated basis for purposes of allocating resources and evaluating financial performance. Accordingly, management has determined that the Company operates in one reportable segment.
| S. | Comprehensive Loss |
The purpose of reporting comprehensive income is to report a measure of all changes in equity of an entity that result from recognized transactions and other economic events of the period resulting from transactions from non-owner sources.
| T. | Leases |
Under Accounting Standards Update, “Leases” (“ASC 842”), the Company determines if an arrangement is a lease at inception. Upon initial recognition, the Company recognizes a liability at the present value of the lease payments to be made over the lease term, and concurrently recognizes a right-of-use asset at the same amount of the liability, adjusted for any prepaid or accrued lease payments, plus initial direct costs incurred in respect of the lease. The Company uses its incremental borrowing rate based on the information available at the commencement date to determine the present value of the lease payments. The subsequent measurement depends on whether the lease is classified as a finance lease or an operating lease. During the reporting periods, the Company has only operating leases. Lease terms include options to extend the lease when it is reasonably certain that the Company will exercise that option. Lease expenses for operating leases are recognized on a straight-line basis over the lease term.
The Company has made a policy election not to capitalize leases with a term of 12 months or less.
In accordance with ASC 360-10, management reviews operating lease assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable based on estimated future undiscounted cash flows. If so indicated, an impairment loss would be recognized for the difference between the carrying amount of the asset and its fair value.
| U. | Recently adopted accounting pronouncements |
From time to time, new accounting pronouncements are issued by FASB, or other standard setting bodies and adopted by the Company as of the specified effective date. Unless otherwise discussed, the impact of recently issued standards that are not yet effective will not have a material impact on our financial position or results of operations upon adoption.
| F-15 |
GALMED PHARMACEUTICALS LTD.
Notes to Consolidated Financial Statements
Note 2 – Significant Accounting Policies (Cont.)
| U. | Recently adopted accounting pronouncements (Cont.) |
In December 2019, the FASB issued ASU 2019-12, “Simplifying the Accounting for Income Taxes”, which will simplify the accounting for income taxes to improve consistency of accounting methods and remove certain exceptions. The amendment is effective for the Company beginning January 1, 2021. The adoption did not have a significant impact on the Company’s consolidated financial statements.
| V. | Recently issued accounting pronouncements |
In May 2021, the FASB issued Update 2021-04, Earnings Per Share (Topic 260), Debt—Modifications and Extinguishments (Subtopic 470-50), Compensation—Stock Compensation (Topic 718), and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40): Issuer’s Accounting for Certain Modifications or Exchanges of Freestanding Equity-Classified Written Call Options (a consensus of the FASB Emerging Issues Task Force). The amendments in this Update affect all entities that issue freestanding written call options that are classified in equity. Specifically, the amendments affect those entities when a freestanding equity-classified written call option is modified or exchanged and remains equity classified after the modification or exchange. The amendments that relate to the recognition and measurement of EPS for certain modifications or exchanges of freestanding equity-classified written call options affect entities that present EPS in accordance with the guidance in Topic 260, Earnings Per Share. The effect of implementing this ASU is immaterial.
| F-16 |
GALMED PHARMACEUTICALS LTD.
Notes to Consolidated Financial Statements
Note 3 – Marketable debt securities
The following table summarizes the Company’s marketable debt securities as of December 31, 2021 and 2020.
| As of December 31, 2021 | ||||||||||||||||
| Gross | Gross | |||||||||||||||
| Amortized | Unrealized | Unrealized | Estimated | |||||||||||||
| Cost | Gains | Losses | Fair Value | |||||||||||||
| (in thousands) | ||||||||||||||||
| Corporate bonds | $ | $ | $ | ( | ) | $ | ||||||||||
| Mutual funds | ( | ) | ||||||||||||||
| Total short-term investments | $ | $ | $ | ( | ) | $ | ||||||||||
| As of December 31, 2020 | ||||||||||||||||
| Gross | Gross | |||||||||||||||
| Amortized | Unrealized | Unrealized | Estimated | |||||||||||||
| Cost | Gains | Losses | Fair Value | |||||||||||||
| (in thousands) | ||||||||||||||||
| Corporate bonds | $ | $ | $ | ( | ) | $ | ||||||||||
| Commercial papers | $ | $ | ( | ) | $ | |||||||||||
| Total short-term investments | $ | $ | $ | ( | ) | $ | ||||||||||
The contractual maturity of the aforementioned marketable securities varies between less than one year to two years.
Note 4– Other Accounts Receivable
| As of December 31, | ||||||||
| 2021 | 2020 | |||||||
| (in thousands) | ||||||||
| Prepaid expenses | $ | $ | ||||||
| Government institutions | ||||||||
| $ | $ | |||||||
Note 5– Leases
The
Company leases, approximately
The
Company elected not to exercise its options to extend the lease and in March 2021, signed a new lease extension agreement to its corporate
headquarters for a period of additional years until March 22, 2023 with an optional one-year renewal period. According to the updated
lease agreement, the aggregate quarterly rental payment for the lease period, together with adjustments and maintenance fees, is approximately
NIS
In addition, the Company leases vehicles under various operating lease agreements.
| F-17 |
GALMED PHARMACEUTICALS LTD.
Notes to Consolidated Financial Statements
Note 5 – Leases (Cont.)
At
December 31, 2021, the Company’s operating lease assets and lease liabilities (both the current and non-current portion) for operating
leases totaled $
The
Company uses its incremental borrowing rate as the discount rate for its leases, as the implicit rate in the lease is not readily determinable.
As of December 31, 2021, the Company’s operating leases had a weighted average remaining lease term of
The following table summarizes the Company’s significant contractual lease obligations at December 31, 2021:
| Less than | ||||||||||||
| Total | 1 year | 1-3 years | ||||||||||
| (in thousands) | ||||||||||||
| Facility leases | $ | $ | $ | |||||||||
| Car leases | ||||||||||||
| Total | $ | $ | $ | |||||||||
Note 6 – Property and equipment, net
| As of December 31, | ||||||||
| 2021 | 2020 | |||||||
| (in thousands) | ||||||||
| Medical equipment | $ | $ | ||||||
| Office furniture and equipment | ||||||||
| Computer software and electronic equipment | ||||||||
| Leasehold improvements | ||||||||
| Less - Accumulated depreciation | ||||||||
| Net book value | $ | $ | ||||||
| F-18 |
GALMED PHARMACEUTICALS LTD.
Notes to Consolidated Financial Statements
Note 7 – Related Parties
| 1. | As
of December 31, 2021, and 2020, the Company had an accrual in the amount of approximately $ | |
| 2. | During
2021, 2020 and 2019, the Company recorded salary expenses, stock-based compensation expenses and directors’ fee to its related
parties in the amount of $ |
Note 8 – Commitments and Contingencies
| 1. | As
of December 31, 2021, the Company recorded a pledge on its short-term deposit in favor of its bank in the amount of approximately
$ | |
| 2. | The Company enters into contracts in the ordinary course of business with contract research organizations for clinical trials and clinical supply manufacturing and with vendors for non-clinical research studies and other services and products for operating purposes, which generally provide for termination upon 30 to 90 days’ notice or less, and therefore are cancelable contracts and not considered as commitment or purchase obligations. | |
| 3. | For information regarding the Company’s leases commitments, see note 5. | |
| 4. | On
June 28, 2021, the Company entered into a license agreement with Yissum Research Development Company of the Hebrew University of
Jerusalem (“Yissum”) pursuant to which Yissum granted to the Company a worldwide, exclusive and irrevocable license to
develop and commercialize Amilo-5Mer. Under the license agreement, the Company is responsible for carrying out the development and
commercialization of Amilo-5Mer and the prosecution and maintenance of the licensed patents under the license agreement. In consideration
for the grant of the license, the Company paid Yissum an upfront license fee of $ | |
| 5. | Other than as described above, the Company did not have any material commitments, including any anticipated material acquisition of plant and equipment or interests in other companies, as of December 31, 2021 and 2020. |
| F-19 |
GALMED PHARMACEUTICALS LTD.
Notes to Consolidated Financial Statements
| A. | Ordinary shares |
| 1. | Ordinary shares confer upon the holders the right to receive notice to participate and vote in general meetings of the Company and the right to receive dividends, if declared. | |
| 2. | On
December 22, 2017, the Company entered into an At-the-Market Equity Offering Sales Agreement (the “Stifel Sales Agreement”)
with Stifel, Nicolaus & Company, Incorporated, as the Company’s sales agent (“Stifel”). Pursuant to the prospectus
relating to the Company’s shelf registration statement on Form F-3 filed with the SEC on March 26, 2018 the Company may offer
and sell, from time to time through Stifel, its ordinary shares having an aggregate offering price of up to $ million. On May 15,
2020, the Company amended and restated the Sales Agreement dated December 22, 2017 between the Company and Stifel, Nicolaus &
Company, Incorporated to include Cantor Fitzgerald & Co. as an additional sales agent for the Company’s “at the market
offering” program. Pursuant to a prospectus supplement filed with the SEC on May 15, 2020, the Company may offer and sell up
to $ million of its ordinary shares. In 2020 the Company sold ordinary shares under the ATM Program for total net proceeds
of approximately $ | |
| 3. | During
February 2021, the Company entered into an underwriting agreement (the “Underwriting Agreement”) with Cantor Fitzgerald
& Co. (the “Underwriter”) in connection with an underwritten public offering (the “Underwritten Public Offering”)
of ordinary shares (the “Firm Shares”) of the Company (the “Ordinary Shares”). The Underwriter
agreed to purchase the Firm Shares from the Company at a price of $ per share. The net proceeds to the Company were approximately
$ | |
| Under
the terms of the Underwriting Agreement, the Company granted the Underwriter an option, exercisable for | ||
| 4. | On March 26, 2021, the Company entered into a new Sales Agreement with Cantor Fitzgerald & Co. and Canaccord Genuity LLC, as sales agents, pursuant to which the Company may offer and sell ordinary shares “at the market” having an aggregate offering price of up to $ million from time to time through the sales agents. | |
| 5. | In November 2021, the Company issued an additional ordinary shares to one of its consultants. |
| F-20 |
GALMED PHARMACEUTICALS LTD.
Notes to Consolidated Financial Statements
Note 9– Shareholders’ Equity (Cont.)
| B. | Stock-based compensation |
| 1. | The Company has an equity-based incentive plan, the 2013 Incentive Share Option Plan (the “2013 Plan”). As of December 31, 2021, a total of shares were reserved for issuance under the 2013 Plan. The 2013 Plan, which was adopted by the Board on September 2, 2013, and approved by the Company’s shareholders on December 30, 2013 (as was amended by the Board and the Company’s shareholders on March 30, 2015, May 11, 2015, and August 30, 2018 respectively), provides for the grant of options to purchase the ordinary shares and the issuance of restricted stock units (“RSUs”) to the Company’s officers, directors, employees, service providers and consultants. The 2013 Plan provides for such equity-based compensation under various and different tax regimes. | |
| 2. | During
the year ended December 31, 2020, certain current and former office holders exercised options into ordinary shares of the
Company for total consideration of $ | |
| 3. | During the year ended December 31, 2020, restricted stock units held by certain officers, employees and former employees vested resulting in the issuance of ordinary shares of the Company. | |
| 4. | In March 2020, the Company granted options to purchase ordinary shares of the Company to several employees. The options are exercisable at $ per share, have a -year term and vest over a period of . The aggregate grant date fair value of such options was approximately $ million. | |
| 5. | In August 2020, the Company granted options to purchase ordinary shares of the Company to a director. The options are exercisable at $ per share, have a -year term and vest over a period of . The aggregate grant date fair value of such options was approximately $ million. In addition, at the Company’s annual shareholder meeting, the shareholders approved a grant of options to purchase ordinary shares of the Company to the Company’s chief executive officer, that was granted by the Company’s board in December 2019. The options are exercisable at $ per share, have a -year term and vest over a period of . The aggregate grant date fair value of such options was approximately $ million. | |
| 6. | In November 2020, the Company granted options to purchase ordinary shares of the Company to several employees. The options are exercisable at $ per share, have a -year term and vest over a period of . The aggregate grant date fair value of such options was approximately $ million. In addition, the Company’s board approved a grant of options to purchase ordinary shares of the Company to Company’s chief executive officer subject to shareholders’ approval (which was obtained in August 2021). The options are exercisable at $ per share, have a -year term and vest over a period of . The aggregate grant date fair value of such options was approximately $ million. | |
| 7. | During
February 2021, certain office holders exercised options into Ordinary shares of the Company for a total amount
of less than $ | |
| 8. | In March 2021, the Company granted options to purchase ordinary shares of the Company to an employee and a consultant. The options are exercisable at $ per share, have a -year term and vest over a period of . The aggregate grant date fair value of such options was approximately $ million. | |
| 9. | In July, 2021, the Company granted options to purchase ordinary shares of the Company to its non-management directors subject to shareholders’ approval (which was obtained in August 2021). The options are exercisable at $ per share, have a -year term and vest over a period of . The aggregate grant date fair value of such options was approximately $ million. | |
| 10. | In August, 2021, the Company granted options to purchase ordinary shares of the Company to certain service providers. The options are exercisable at $ per share, have a -year term and vest over a period of . The aggregate grant date fair value of such options was approximately $ million. |
| F-21 |
GALMED PHARMACEUTICALS LTD.
Notes to Consolidated Financial Statements
Note 9– Shareholders’ Equity (Cont.)
| B. | Stock-based compensation (Cont.) |
| 7. | A summary of the status of the Company’s option plans as of December 31, 2021 and 2020 and changes during the years then ended are presented below: |
| 2021 | 2020 | |||||||||||||||
| Weighted | Weighted | |||||||||||||||
| Number of | average | Number of | average | |||||||||||||
| share options | exercise price | share options | exercise price | |||||||||||||
| Options outstanding at beginning of year | $ | $ | ||||||||||||||
| Granted | $ | $ | ||||||||||||||
| Forfeited | ( | ) | $ | ( | ) | $ | ||||||||||
| Exercised | ( | ) | $ | ( | ) | $ | ||||||||||
| Outstanding at end of year | $ | $ | ||||||||||||||
| Options exercisable at year end | $ | $ | $ | |||||||||||||
The following assumptions were used for the fiscal year 2021, 2020 and 2019 grants:
| - | dividend yield of % for all periods. | |
| - | risk-free interest rate between % and %for the fiscal year 2019, % and % for the fiscal year 2020 and % and % for the fiscal year 2021. | |
| - | an expected life between and years for all periods. | |
| - | and a volatility rate ranging between % and % for the fiscal year 2019; and % and % for the fiscal year 2020 and % and % for the fiscal year 2021. |
As of December 31, 2021, and 2020, the weighted-average remaining contractual term of the outstanding options, excluding the options granted in 2002 that have no expiration date, is and years, respectively.
The weighted average grant date fair value of the options granted during the years ended December 31, 2021, 2020 and 2019 is $, $, and $ respectively.
As of December 31, 2021, a total of the outstanding and exercisable options are “in the money” with aggregate intrinsic value of $ million; while as of December 31, 2020 a total of outstanding and exercisable options were “in the money” with aggregate intrinsic value of $ million.
The unrecognized compensation expense calculated under the fair-value method for stock options expected to vest as of December 31, 2021, 2020 and 2019 is approximately $ million, $ million, and $ million, respectively, and is expected to be recognized over a weighted-average period of years, years and years, respectively.
For the years ended 2021, 2020 and 2019, the Company recorded a total of $ million, $ million, and $ million of stock-based compensation expenses, in connection with the above-mentioned options.
| F-22 |
GALMED PHARMACEUTICALS LTD.
Notes to Consolidated Financial Statements
Note 9– Shareholders’ Equity (Cont.)
| B. | Stock-based compensation (Cont.) |
During 2016, the Company issued a total of restricted stock units (“RSU”). Upon vesting, each RSU will settle by the issuance of one ordinary share. The RSUs vest over . As of December 31, 2021, a total of ordinary shares were issued upon vesting and there are no outstanding RSUs., For the years 2021, 2020 and 2019, with respect to the above-mentioned RSUs, the Company recorded stock-based compensation expenses in the amount of $, $ thousand and $ thousand, respectively. All of the above-mentioned stock-based compensation expenses are recorded under general and administrative expenses. as of December 31, 2021, there are outstanding RSU’s and unrecognized compensation expense.
Note 10 – Research and Development Expenses
| Year ended December 31, | ||||||||||||
| 2021 | 2020 | 2019 | ||||||||||
| (in thousands) | ||||||||||||
| Chemistry and formulation studies | $ | $ | $ | |||||||||
| Salaries | ||||||||||||
| Stock-based compensation | ||||||||||||
| Research and preclinical studies | ||||||||||||
| Clinical studies | ||||||||||||
| Regulatory and other expenses | ||||||||||||
| $ | $ | $ | ||||||||||
Note 11 – General and Administrative Expenses
| Year ended December 31, | ||||||||||||
| 2021 | 2020 | 2019 | ||||||||||
| (in thousands) | ||||||||||||
| Stock-based compensation | $ | $ | $ | |||||||||
| Professional fees | ||||||||||||
| Salaries and benefits | ||||||||||||
| Rent and office-maintenance fees | ||||||||||||
| Investor relations and business development expenses | ||||||||||||
| Insurance and other expenses | ||||||||||||
| $ | $ | $ | ||||||||||
| F-23 |
GALMED PHARMACEUTICALS LTD.
Notes to Consolidated Financial Statements
Note 12 – Financial income, net
| Year ended December 31, | ||||||||||||
| 2021 | 2020 | 2019 | ||||||||||
| (in thousands) | ||||||||||||
| Bank fees | $ | $ | $ | |||||||||
| Interest income | ( | ) | ( | ) | ( | ) | ||||||
| Loss (gain) from sale of marketable debt securities | ( | ) | ( | ) | ||||||||
| Foreign currency losses | ||||||||||||
| $ | ( | ) | $ | ( | ) | $ | ( | ) | ||||
Note 13 – Income Taxes
| A. | General |
The Company is assessed for tax purposes on an unconsolidated basis. Each of the Company’s subsidiaries is subject to the tax rules prevailing in its country of incorporation.
| B. | Corporate Taxation Israeli Companies: |
Ihe
Israeli corporate income tax rate is
On
February 7, 2018, the Israeli Tax Authority issued a ruling granting the Company’s Israeli subsidiary, Galmed Research and Development
Ltd, a “Preferred Technological Enterprise” status as defined under the Encouragement of Capital Investment Law -1959 (the
“Approval”). The grant of the status means that the Company’s Israeli subsidiary will be subject to a reduced Israeli
corporate tax rate that will range between
Maltese subsidiary:
Taxable
income of Maltese companies was subject to tax at the rate of
| C. | Net Operating Loss Carry forward |
As
of December 31, 2021, the Company had approximately $
| F-24 |
GALMED PHARMACEUTICALS LTD.
Notes to Consolidated Financial Statements
Note 13 – Income Taxes (Cont.)
| D. | Deferred income taxes |
As
of December 31, 2021, the significant components of the Company’s deferred tax assets are net operating loss carryforward in the
amount of $
| E. | Tax assessments |
The Israeli subsidiaries received final tax assessments through the year ended December 31, 2016.
| F. | Effective tax expense |
A reconciliation of the Company’s effective tax expense to the Company’s theoretical statutory tax benefit is as follows:
| Year ended December 31, | ||||||||||||
| 2021 | 2020 | 2019 | ||||||||||
| (in thousands) | ||||||||||||
| Loss before taxes on income, as reported in the consolidated statements of operations | $ | $ | $ | |||||||||
| Statutory tax rate | % | % | % | |||||||||
| Theoretical tax benefit | ||||||||||||
| Losses and other items for which a valuation allowance was provided or benefit from loss carry forwards | ( | ) | ( | ) | ( | ) | ||||||
| Actual tax expense | $ | $ | $ | |||||||||
| F-25 |
ITEM 19. Exhibits.
| 162 |
| 101 | The following financial information from Galmed Pharmaceuticals Ltd.’s Annual Report on Form 20-F for the year ended December 31, 2021, formatted in Inline Extensible Business Reporting Language (XBRL): (i) Consolidated Balance Sheets, (ii) Consolidated Statements of Operations, (iii) Consolidated Statements of Comprehensive Loss, (iii) Consolidated Statements of Changes in Shareholders’ Equity (iii) the Consolidated Statements of Cash Flows, and (iv) Notes to Consolidated Financial Statements | |
| 101.INS | Inline XBRL Instance Document | |
| 101.SCH | Inline XBRL Taxonomy Extension Schema Document | |
| 101.CAL | Inline XBRL Taxonomy Extension Calculation Linkbase Document | |
| 101.DEF | Inline XBRL Taxonomy Extension Definition Linkbase Document | |
| 101.LAB | Inline XBRL Taxonomy Extension Label Linkbase Document | |
| 101.PRE | Inline XBRL Taxonomy Extension Presentation Linkbase Document | |
| 104 | Cover Page Interactive Data File (embedded within the Inline XBRL document) |
| (1) | Incorporated herein by reference to Amendment No. 1 to the Registration Statement on Form F-1, filed with the SEC on February 28, 2014. | |
| (2) | Incorporated herein by reference to the Registration Statement on Form F-1, filed with the SEC on February 6, 2014. | |
| (3) | Incorporated herein by reference to the Company’s Report on Form 6-K filed with the SEC on June 1, 2016. | |
| (4) | Incorporated herein by reference to Exhibit A to the Company’s Report on Form 6-K filed with the SEC on April 2, 2015. | |
| (5) | Incorporated herein by reference to Annex A to the Company’s Report on Form 6-K filed with the SEC on July 8, 2020. | |
| (6) | Incorporated herein by reference to the Company’s Annual Report on Form 20-F filed with the SEC on March 31, 2015. | |
| (7) | Incorporated herein by reference to the Company’s Annual Report on Form 20-F filed with the SEC on March 22, 2016. | |
| (8) | Incorporated herein by reference to the Company’s Annual Report on Form 20-F filed with the SEC on March 23, 2017. | |
| (9) | Incorporated herein by reference to Exhibit 1.1 to the Company’s Report on Form 6-K filed with the SEC on March 26, 2021. | |
| (10) | Incorporated herein by reference to the Company’s Annual Report on Form 20-F filed with the SEC on March 13, 2018. | |
| (11) | Incorporated herein by reference to Exhibit 1.1 to the Company’s Report on Form 6-K filed with the SEC on February 18, 2021. | |
| (12) | Incorporated herein by reference to the Company’s Annual Report on Form 20-F filed with the SEC on March 13, 2019. | |
| (13) | Incorporated herein by reference to the Company’s Annual Report on Form 20-F filed with the SEC on March 12, 2020. | |
| (14) | Incorporated herein by reference to the Company’s Annual Report on Form 20-F filed with the SEC on March 18, 2021. |
| * | Filed herewith. |
| 163 |
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
| GALMED PHARMACEUTICALS LTD. | ||
| By: | /s/ Allen Baharaff | |
| Allen Baharaff | ||
| President, Chief Executive Officer and Chairman | ||
| Date: May 2, 2022 | ||
| 164 |
Exhibit 1.1
THE COMPANIES LAW, 5759-1999
AMENDED AND RESTATED ARTICLES OF ASSOCIATION
of
GALMED PHARMACEUTICALS LTD
גלמד פרמסוטקלס בע”מ
Preliminary
| 1. | (a) | In these Articles, unless the context otherwise requires: |
| (a) | An “Annual General Meeting” shall have the meaning ascribed to such term in Article 50 hereunder. |
| (b) | The “Board of Directors” or “Board” means the board of directors of the Company. |
| (c) | A “Board Meeting” means a meeting of the Board of Directors. |
| (d) | The “Companies Law” means the Companies Law, 5759-1999 or any law which may replace or amend it, as shall be in force from time to time. |
| (e) | The “Company” means Galmed Pharmaceuticals Ltd. |
| (f) | A “Special General Meetings” shall have the meaning ascribed to such term in Article 50 hereunder. |
| (g) | An “Extraordinary Transaction” shall have the meaning ascribed to such term in the Companies Law. |
| (h) | A “General Meeting” means a meeting of the Shareholders, whether an Annual General Meeting or a Special General Meeting, both as defined herein. |
| (i) | The “Office” means the registered office of the Company for the time being. |
| (j) | An “Office Holder” shall have the meaning ascribed to such term in the Companies Law. |
| (k) | The “Register” means the principal register of Shareholders specified in Article 93, to be kept in accordance with the Companies Law, and/or, if the Company shall have any additional or branch register(s), any such additional or branch register(s) as the case may be. |
| (l) | The “Restrictive Trade Law” means the Israel Restrictive Trade Practices Law, 5748-1988. |
| (m) | The “SEC” means the United States Securities and Exchange Commission. |
| (n) | The “Securities Law” means the Israel Securities Law, 5728-1968. |
| (o) | A “Shareholder” means any person registered in the Register as the owner of shares of the Company. |
| - 2 - |
| (p) | A “Shareholder Resolution” means a resolution adopted by a simple majority of the voting rights of the Company represented, personally or by proxy, and voting with respect thereto, unless a different majority is required in respect to such matter pursuant to the Companies Law or these Articles at the time the resolution is voted on, in which case a “Shareholder Resolution” shall mean a resolution adopted by such required majority. |
| (b) | Subject to the provisions of this Article, in these Articles, unless the context otherwise requires, words and expressions used herein which are defined in the Companies Law, or any modification thereof in force at the date at which these Articles become binding upon the Company, shall have the meaning so defined, words importing the singular shall include the plural and vice versa, words importing the masculine gender shall include the feminine and neuter genders and vice versa, words importing persons shall include bodies corporate and the captions used herein shall not be deemed to affect the construction of any provision hereof. |
| (c) | The Company may donate reasonable amounts to any worthy cause, as determined and approved by the Board of Directors, even if such donation is not made for business considerations. |
Purpose
| 2. | The purpose of the Company is to engage in any lawful activity. |
Limited Liability
| 3. | The liability of each Shareholder is limited to the unpaid sum, if any, owing to the Company in consideration for the issuance of the shares of the Company held by such shareholder. |
Share Capital
| 4. | (a) | The authorized share capital of the Company is 500,000 (five hundred thousand) New Israeli Shekels (“NIS”), divided into 50,000,000 (fifty million) Ordinary Shares of NIS 0.01 (one Agora) each, all ranking pari passu (“Ordinary Shares”). |
| (b) | Ordinary Shares in respect of which all calls have been fully paid shall confer on their holders the right to attend and to vote at General Meetings of the Company. Subject to the rights of holders of shares with limited or preferred rights, Ordinary Shares shall confer upon the holders thereof equal rights to receive dividends and to participate in the distribution of the assets of the Company upon its winding-up, in proportion to the amount paid up or credited as paid up on account of the nominal value of the shares held by them respectively and in respect of which such dividends are being paid or such distribution is being made, without regard to any premium paid in excess of the nominal value, if any. |
Shares
| 5. | Without prejudice to any special rights previously conferred upon the holders of existing shares of the Company, the Company may, from time to time, by Shareholder Resolution, provide for shares with such preferred or deferred rights or rights of redemption or other special rights and/or such restrictions, whether in regard to dividends, voting, repayment of share capital or otherwise, as may be stipulated in such Shareholder Resolution. |
| 6. | (a) | If at any time the share capital is divided into different classes of shares, the Company may by Shareholder Resolution, unless otherwise provided by the terms of issue of the shares of that class, modify, convert, broaden, add or otherwise alter the rights, privileges, advantages, restrictions and provisions related or unrelated at that time to the shares of any class either with the consent in writing of the holders of at least 75% of the issued shares of that class or with the sanction of a resolution passed by a simple majority of those present, personally or by proxy, and voting thereon at a separate general meeting of the holders of the shares of that class. |
| - 3 - |
| (b) | The provisions of these Articles relating to General Meetings and to the convening thereof and to notices in respect thereof and to resolutions to be passed thereat shall mutatis mutandis apply to every separate general meeting as mentioned above. |
| (c) | Unless otherwise provided by these Articles, the enlargement of an existing class of shares, or the issuance or allotment of additional shares thereof, or the creation of additional shares of that class as a result of conversion of shares from another class or the unification with another class shall not be deemed to modify or alter the rights attached to the previously issued shares of such class or of any other class. |
| 7. | (a) | The unissued shares shall be under the control of the Board of Directors who may issue or allot them or give any person the option to acquire them or otherwise dispose of them for cash or other consideration to such persons, on such terms and conditions, and either at a premium or at par, or, subject to the provisions of the Companies Law, at a discount and at such times as the Board of Directors may deem fit, and with full authority to serve on any person a call on any shares as provided in Article 15 below, during such time and for such consideration as the Board of Directors may deem fit. |
| (b) | The Company may pay a commission to any person in consideration of his subscribing or agreeing to subscribe, whether absolutely or conditionally, for any shares or other securities in the Company, or procuring or agreeing to procure subscriptions, whether absolute or conditional, for any shares or other securities in the Company, as the Board of Directors may deem fit. Such commission may be paid in cash or in fully or partly paid shares of the Company, or in a combination of such methods. |
| 8. | If by the conditions of allotment of any share, the whole or any part of the price thereof shall be payable by installments, every such installment shall, when due, be paid to the Company by the registered holder of the share for the time being or from time to time or by his administrators. |
| 9. | (Reserved). |
| 10. | Save as herein otherwise provided, the Company shall be entitled to treat the registered holder of any share as the absolute owner thereof, and, accordingly, shall not, except as ordered by a court of competent jurisdiction, or as by statute required, be bound to recognize any equitable or other claim to or interest in such share on the part of any other person and the Company shall not be bound by or required to recognize any equitable, contingent, future or partial interest in any shares or any right whatsoever in respect of any shares other than an absolute right to the entirety thereof in the registered holder. |
Share Certificates
| 11. | The certificates of title to shares (“Share Certificates”) shall be issued under the seal or the rubber stamp of the Company and shall bear the manual or facsimile signatures of two Directors, or one Director and the Secretary of the Company, or such other persons as are authorized by the Board of Directors. In case any Director, officer or such other authorized person who has signed or whose facsimile signature has been placed upon a share certificate shall have ceased to be such Director, officer or authorized person before such certificate is issued, it may be issued by the Company with the same effect as if he were such Director, officer or authorized person at the date of issue. |
| 12. | Every Shareholder shall be entitled without payment to receive one Share Certificate representing in aggregate all the shares registered in his name. |
| 13. | Share Certificates of shares registered in the names of two or more persons shall be delivered to the person first named in the Register in respect of such co-ownership and such delivery shall be deemed sufficient delivery to all co-owners. The Company shall not be bound to issue more than one Share Certificate to the joint holders. |
| - 4 - |
| 14. | If a Share Certificate is defaced, lost or destroyed, it may be renewed upon production of such evidence of loss, the provision of such indemnities and the payment of such handling fee (if any) as the Board of Directors thinks fit. |
Calls
| 15. | The Board of Directors may from time to time make such calls as it deems fit upon the Shareholders in respect of all moneys unpaid on the shares held by them respectively, and by the conditions of allotment thereof not made payable at fixed times, and each Shareholder shall pay the amount of every call so made on him to the persons and at the time and place appointed by the Board of Directors. A call may be made payable by installments and shall be deemed to have been made when the resolution of the Board of Directors authorizing such call is passed. |
| 16. | At least fourteen days’ notice of any call shall be given, specifying the time and place of payment, and to whom such call shall be paid, provided that before the time for payment of such call the Board of Directors may, by notice in writing to the Shareholders, revoke the same or extend the time for payment thereof. |
| 17. | The joint holders of a share shall be jointly and severally liable to pay all calls in respect thereof. |
| 18. | If by the terms of issue of any share or otherwise any amount is made payable at any fixed time or by installments at fixed times, whether on account of the nominal value of the share or by way of premium, every such amount or installment shall be payable as if it were a call duly made by the Board of Directors of which due notice had been given, and all the provisions herein contained in respect of such calls shall apply to such amount or to such installment. |
| 19. | If the amount of any call or installment is not paid on or before the due date for payment thereof, then the person who is for the time being the owner of the share on which the call was made or the installment became due shall pay interest on the said amount at the maximum rate permissible under law for the time being, or at such lesser rate as may be fixed by the Board of Directors from time to time, as from the date for payment until the same is actually paid. The Board of Directors shall, however, be at liberty to waive the payment of interest, wholly or in part. No Shareholder shall be entitled to receive any dividend or to exercise any privileges as a Shareholder until he shall have paid all calls for the time being due and payable on every share held by him whether alone or jointly with any other person together with interest and expenses (if any). |
| 20. | If the Board of Directors deems fit, it may receive from any Shareholder willing to advance the same, any amounts due on account of all or any of his shares which have not yet been called or in respect of which the date of payment has not yet fallen due, and, unless otherwise agreed with such Shareholder, the Board of Directors may pay him interest on all or any of the amounts so advanced, up to the date when the same would, if not paid in advance, have fallen due, at such rate of interest as may be agreed upon between the Board of Directors and such Shareholder, and the Board of Directors may at any time repay any amount so advanced by giving such Shareholder seven days’ prior notice in writing. |
| 21. | The Board of Directors may determine differences between Shareholders in relation to the amount of any call and to the date of payment. |
Forfeiture and Lien
| 22. | If any Shareholder fails to pay any call or installment on or before the day appointed for payment of the same, the Board of Directors may at any time thereafter, as long as the said call or installment remains unpaid, resolve to forfeit all or any of the shares in the event the Shareholder does not pay the same, as provided below, together with any interest that may have accrued and all expenses that may have been incurred by reason of such non-payment. |
| - 5 - |
| 23. | Notice of any such resolution shall be served on the Shareholder. The notice shall specify a day (being not less than 14 days from the date of the notice) and a place or places on and at which such call or installment and such interest and expenses as aforesaid are to be paid. The notice shall also state that in the event of non-payment at or before the time and at the place appointed, the shares in respect of which the call was made or installment is payable will be ipso facto forfeited, save shares that have been fully paid for. |
| 24. | Any forfeiture as aforesaid shall include all dividends declared in respect of the forfeited shares and not actually paid before the forfeiture. |
| 25. | Any share so forfeited shall be the property of the Company, and the Board of Directors may, subject to the provisions hereof, sell, re-allot and otherwise dispose of the same as it may deem fit. |
| 26. | The Board of Directors may, at any time before any share so forfeited shall have been sold, re-allotted or otherwise disposed of, annul the forfeiture on such conditions as it deems fit. No such annulment shall estop the Board of Directors from re-exercising its powers of forfeiture pursuant to these Articles. |
| 27. | Any Shareholder whose shares have been forfeited shall cease to be a Shareholder in respect of the forfeited shares, but shall, notwithstanding, be liable to pay, and shall forthwith pay, to the Company, all calls, installments, interest and expenses owing upon or in respect of such shares at the time of forfeiture, together with interest thereon from the time of forfeiture, until payment, at the maximum rate of interest permissible under law for the time being, and the Board of Directors may enforce the payment of such moneys, or any part thereof, if it so thinks fit, but shall not be under any obligation to do so. |
| 28. | The provisions of these Articles relating to forfeiture shall apply to any case of nonpayment of a known sum which, according to the terms of issue or allotment of the share, is payable at any fixed time, whether on account of the nominal value of the share or by way of premium, as if such sum were payable under a call duly made, notified and delivered. |
| 29. | Except to the extent that the same may be waived or subordinated in writing, the Company shall have a first and paramount lien upon all the shares registered in the name of each Shareholder (without regard to any equitable or other claim or interest in such shares on the part of any other person), and upon the proceeds of the sale thereof, for his debts, liabilities and obligations to the Company arising from any amount payable by such Shareholder in respect of any unpaid or partly paid share, whether or not such debt, liability or obligation has matured. Such lien shall extend to all dividends from time to time declared or paid in respect of such share. Unless otherwise decided by the Board, the registration by the Company of a transfer of shares shall be deemed to be a waiver on the part of the Company of the lien (if any) on such shares, immediately prior to such transfer. |
| 30. | For the purpose of enforcing such lien, the Board of Directors may sell the shares subject thereto in such manner as it deems fit; but no sale shall be made until the time for the fulfillment or discharge of the debts, liabilities and engagements as aforesaid shall have arrived, and until notice in writing of the Company’s intention to sell shall have been served on such Shareholder, his executors or administrators, and the payment, fulfillment or discharge of such debts, liabilities or engagements shall not have been made during the seven days after such notice. |
| 31. | The net proceeds of any such sale, after payment of the costs thereof, shall be applied in or towards satisfaction of the debts, liabilities or engagements of such Shareholder (including debts, liabilities and engagements which have not yet fallen due for payment or satisfaction) and the remainder (if any) shall be paid to the Shareholder, his executors, administrators or assigns. |
| 32. | Upon any sale after forfeiture or for enforcing a lien in exercise of the powers hereinbefore given, the Board of Directors may appoint some person to execute an instrument of transfer of the shares sold and cause the purchaser’s name to be entered in the Register in respect of the shares sold, and the purchaser shall not be bound to see to the regularity of the proceedings, or to the application of the purchase money, and after his name has been entered in the Register in respect of such shares, the validity of the sale shall not be impeached by any person, and the remedy of any person aggrieved by the sale, if grounds for any remedy exist in accordance with law, shall be in damages only and against the Company exclusively. |
| - 6 - |
Transfer and Transmission of Shares
| 33. | (a) | Any transfer of shares of the Company which have not been fully paid-up will be subject to the approval of the Board of Directors. The Board of Directors may, at its sole discretion, refuse to approve a transfer of shares as aforesaid, without the requirement to provide reasons for its decision. |
| (b) | The transfer of shares which have been fully paid-up is not subject to the approval of the Board of Directors. |
| 34. | No transfer of shares shall be registered or, if such approval is required, approved by the Board of Directors unless a proper instrument of transfer has been submitted to the Company (or its transfer agent) together with the Share Certificate for the transferred shares (if such has been issued) and with any other evidence the Board of Directors may require in order to prove to its satisfaction the rights of the intending transferor in the transferred shares. |
| 35. | (a) | The instrument of transfer shall be signed by the transferor and the transferee, and the transferor shall be considered by the Company as the owner of the shares until the transferee is registered in the Register in respect of the shares transferred to him. The Board may decide that, with respect to a transfer of fully paid-up shares, the instrument of transfer need only be signed by the transferor. The Board may also decide that the signature of a witness on the instrument of transfer is not necessary. The instrument of transfer of any share shall be in writing in such usual or accepted form or forms as shall be approved by the Board of Directors. |
| (b) | The Company may impose a fee for registration of a share transfer, at such reasonable rate as may be determined by the Board from time to time. |
| 36. | Instruments of transfer that are registered shall remain in the Company’s possession; however, instruments of transfer which the Board of Directors refuses to register in accordance with Article 33(a), 34 or 35 above shall, on demand made by whomever delivered them, be returned to such person together with the Share Certificate (if delivered). |
| 37. | The executors and administrators of a deceased sole holder of a share, or, if there are no executors or administrators, the persons beneficially entitled as heirs of a deceased sole holder, shall be the only persons recognized by the Company as having any title to the share. In case of a share registered in the names of two or more holders, the Company shall recognize the survivor or survivors as the only persons having any title to or benefit in the share. Nothing herein contained shall release the estate of a deceased joint holder from any liability in respect of any share jointly held by him. |
| 38. | Any person becoming entitled to a share in consequence of the death of any person, upon producing evidence of the grant of probate or letters of administration or declaration of succession or such other evidence as the Board of Directors may deem sufficient that he sustains the character in respect of which he proposes to act under this Article or of his title, shall be registered as a Shareholder in respect of such shares, or may, subject to the regulations as to transfer herein contained, transfer such shares. |
| 39. | The Company may recognize the receiver or liquidator of any Shareholder in winding-up or dissolution, or the trustee in bankruptcy or any official receiver of a bankrupt Shareholder, as being entitled to the shares registered in the name of such Shareholder. |
| - 7 - |
| 40. | The receiver or liquidator of a Shareholder in winding-up or dissolution, or the trustee in bankruptcy or any official receiver of any bankrupt Shareholder, upon producing such evidence as the Board of Directors may deem sufficient that he sustains the character in respect of which he proposes to act under this Article or of his title, may, with the consent of the Board of Directors (which the Board of Directors may refuse to grant without giving any reason for its refusal), be registered as a Shareholder in respect of such shares, or may, subject to the regulations as to transfer herein contained, transfer such shares. |
| 41. | A person upon whom the ownership of a share devolves by transmission shall be entitled to receive, and may give a discharge for, any dividends or other monies payable in respect of the share but he shall not be entitled in respect of it to receive notices, or to attend or vote at General Meetings of the Company, or, save as otherwise provided herein, to exercise any of the rights or privileges of a Shareholder, unless and until he shall be registered in the Register. |
Redeemable Shares
| 42. | The Company may, subject to the provisions of the Companies Law, issue redeemable shares and redeem them. |
Alteration and Increase of Share Capital
| 43. | The Company may from time to time, by Shareholder Resolution, whether or not all the shares authorized have been issued, and whether or not the whole of the shares then issued has been called up for payment, increase its share capital by the creation of new shares, and such increase shall be in such amount and shall be divided into shares of such nominal amounts, and be issued subject to such restrictions and terms and with such rights and preferences, as the resolution creating the same shall provide. In particular the shares may be issued with preferential or deferred rights as to dividends or the distribution of assets and with special, limited or no voting rights. |
| 44. | Unless otherwise provided in the resolution authorizing the increase of share capital, the new shares shall be subject to the same provisions applicable to the shares of the original capital with regard to the payment of calls, lien, forfeiture, transfer, transmission and otherwise. |
| 45. | The Company may, by Shareholder Resolution and in accordance with and subject to the Companies Law: |
| (a) | consolidate its share capital or any portion thereof and divide it into shares of larger nominal value than its existing shares; |
| (b) | divide its existing shares or any portion thereof by subdivision into shares of smaller nominal value; |
| (c) | cancel any unissued shares provided there is no obligation of the Company, including a contingent obligation, to issue the shares, and reduce in such manner its share capital by the amount of the shares which are cancelled; and/or |
| (d) | reduce its share capital in any manner permitted by law and subject to any condition required by law. |
| 46. | With respect to any consolidation of issued shares into shares of larger nominal value, and with respect to any other action which may result in fractional shares, the Board of Directors may settle any difficulty which may arise with regard thereto as it deems fit, including, inter alia, by means of one or more of the following actions, subject to applicable law: |
| (a) | determine, as to the holder of shares so consolidated, which issued shares shall be consolidated into each share of larger nominal value; |
| - 8 - |
| (b) | allot, in contemplation of or subsequent to such consolidation or other action, such shares or fractional shares sufficient to preclude or remove fractional shareholdings; |
| (c) | redeem in the case of redeemable shares, and subject to applicable law, such shares or fractional shares sufficient to preclude or remove fractional shareholdings; and |
| (d) | cause the transfer of fractional shares by certain Shareholders of the Company to other Shareholders thereof so as to most expediently preclude or remove any fractional shareholdings, and cause the transferees to pay the transferors the fair value of fractional shares so transferred, and the Board of Directors is hereby authorized to act as agent for the transferors and transferees with power of substitution for purposes of implementing the provisions of this Article. |
Purchase of the Company’s Shares
| 47. | The Company may, subject to and in accordance with the provisions of the Companies Law, purchase or undertake to purchase, or provide finance and/or assistance or undertake to provide finance and/or assistance, directly or indirectly, with respect to the purchase of, its shares or securities which may be converted into shares of the Company or which confer rights upon the holders thereof to purchase shares of the Company. |
Borrowing Powers
| 48. | The Board of Directors may from time to time, at its discretion, borrow or secure the payment of any sum or sums of money for the purposes of the Company. The Directors may raise or secure the repayment of such sum or sums in such manner, at such times and upon such terms and conditions in all respects as they think fit and, in particular, by the issue of bonds, perpetual or redeemable debentures, debenture stock or any mortgages, charges or other securities on the undertaking of the whole or any part of the property of the Company, both present and future, including its uncalled capital for the time being and its called but unpaid capital. |
Record Date for General Meetings
| 49. | Notwithstanding any other provision of these Articles to the contrary, and subject to applicable law, the Board of Directors may fix a date, not exceeding 40 days prior to the date of any General Meeting, as the date as of which Shareholders entitled to vote at such meeting shall be determined, and all persons who are registered in the Register as holders of voting shares on such date and no others shall be entitled to vote at such meeting. A determination of Shareholders of record entitled to vote at any General Meeting shall apply to any adjournment of such meeting; provided however, that the Board may fix a new record date for the adjourned meeting. |
General Meetings
| 50. | An annual General Meeting shall be held at least once in every calendar year, not later than 15 months after the last preceding annual General Meeting, at such time and place as the Board of Directors may determine, and such meetings shall be called “Annual General Meetings”. The function of Annual General Meetings shall be to elect Directors in accordance with these Articles, receive and consider the profit and loss account, the balance sheet and the ordinary reports and accounts of the Directors and auditors, appoint auditors and transact any other business which under these Articles or applicable law may be transacted by the shareholders of a company in general meeting. All other General Meetings shall be called “Special General Meetings”. |
| 51. | The Board of Directors may whenever it thinks fit convene a Special General Meeting, and it shall be obliged to do so upon a request in writing as provided in the Companies Law. |
| - 9 - |
| 52. | (a) | The Company shall not be required to deliver or serve notice (‘Hodaa’) of General Meetings or of any adjournments thereof to any Shareholder. |
| (b) | Without derogating from the provisions of Article 52(a) above, the Company will publicize the convening of General Meetings in any manner reasonably determined by the Company, such as by filing an appropriate periodic report with the SEC, by posting a notice on the Company’s website or by publishing in one or more international wire services or in one or more newspapers, and any such publication shall be deemed duly made, given and delivered to all Shareholders on the date on which it is first made, posted, filed or published in the manner so determined by the Company in its sole discretion. |
Proceedings at General Meetings
| 53. | (a) | No business shall be transacted at a General Meeting unless the requisite quorum is present at the commencement of the meeting. Unless otherwise provided in these Articles, two or more Shareholders, present in person or by proxy, holding shares conferring in the aggregate more than 33.33% of the voting rights of the Company on the record date, shall constitute a quorum. |
| (b) | If within half an hour from the time appointed for the General Meeting a quorum is not present, the General Meeting, if convened by the Board upon the demand of Shareholders or upon the demand of less than 50% of the Directors then in office or directly by such Shareholders or Directors, shall be cancelled. Otherwise, if a General Meeting is called and no quorum is present within half an hour from the time appointed for such General Meeting, it shall stand adjourned to the same day in the following week, at the same time and place or to such other day, time and place as the Directors may determine and specify in the publication with respect to the General Meeting. It shall not be necessary to give notice of or publicize such adjournment. If at such adjourned General Meeting a quorum is not present within half an hour from the time stated, any two Shareholders present in person or by proxy shall constitute a quorum even if, between them, they represent shares conferring 33.33% or less of the voting rights of the Company. |
| 54. | Unless otherwise prescribed by applicable law or by these Articles, a resolution of the Shareholders will be deemed adopted if approved at a General Meeting at which a quorum is present by a simple majority of the voting rights of the Company (as set forth in Article 62 below) represented personally or by proxy and voting thereon; provided, however, that a resolution with respect to the amendment or replacement of the Articles of Association of the Company shall require the affirmative vote of at least 75% of the voting rights of the Company represented personally or by proxy and voting thereon at a General Meeting at which a quorum is present. |
| 55. | (a) | The Chairman of the Board of Directors will serve as the chairman of General Meetings of the Company. If such Chairman shall have indicated in advance that he will not be attending, or shall be unwilling to act in such capacity, or shall not be present within 15 minutes from the time stated for the commencement of the meeting, the most senior of the Directors present (such seniority to be determined by the length of time such person has served as a Director) and willing to do so will chair the meeting and, if no Director is present or if no Directors are willing to chair the meeting, those present may choose from amongst themselves a person to chair the meeting. |
| (b) | The chairman of any General Meeting shall not be entitled to a second or casting vote. |
| 56. | Every question submitted to a General Meeting shall be decided by a show of hands, but if a written ballot is demanded by a Shareholder, present in person or by proxy and entitled to vote at the meeting, the same shall be decided by a written ballot. A written ballot may be demanded before the proposed resolution is voted upon or immediately after the declaration by the chairman of the results of the vote by a show of hands. If a written ballot is demanded after such declaration, the results of the vote by a show of hands shall be of no effect and the proposed resolution shall be decided by the written ballot. |
| - 10 - |
| 57. | If a written ballot is demanded as aforesaid, it shall be taken in such manner and at such time and place as the chairman of the General Meeting directs, and either at once or after an interval or adjournment, or otherwise, and the result of the written ballot shall be deemed to be the resolution of the General Meeting in respect of which the written ballot is demanded. The demand for a written ballot may be withdrawn at any time before the written ballot is taken. |
| 58. | (a) | The demand for a written ballot shall not prevent the continuation of the General Meeting for the transaction of any business other than in respect of the question on which the written ballot has been demanded. |
| (b) | A written ballot demanded on the election of a chairman or on a question of an adjournment of a General Meeting shall be taken forthwith. |
| 59. | A declaration by the chairman of the General Meeting that a resolution has been carried unanimously, or carried by a particular majority, or rejected, and an entry to that effect in the book of proceedings of the Company, shall be conclusive evidence of the fact without proof of the number or proportion of the votes recorded in favor of or against such resolution. |
| 60. | The chairman of a General Meeting at which a quorum is present may, with the consent of the holders of a majority of the voting rights of the Company represented, personally or by proxy, at the General Meeting and voting on the question of adjournment, adjourn the same from time to time and from place to place and the chairman shall do so if so directed by the General Meeting; but no business shall be transacted at any adjourned General Meeting other than the business left unfinished at the General Meeting from which the adjournment takes place. The Company will publicly announce the adjournment and the matters to be included on the agenda of the adjourned General Meeting in the same manner in which it announced the convening of the original General Meeting. |
| 61. | Subject to applicable law, a resolution in writing signed by all Shareholders then entitled to vote at General Meetings or to which all such Shareholders have given their written consent (including, but not limited to, by letter, telegram, telex, facsimile, electronic mail or otherwise) shall be deemed to have been adopted as if it were adopted as a Shareholder Resolution at a General Meeting duly convened and held. Any such resolution may consist of several documents in like form and signed or consented to as aforesaid, by one or more Shareholders. |
Votes of Shareholders
| 62. | Subject to any special conditions, rights or restrictions as to voting rights set forth in the terms of issue of any shares or attached at the time to any class of shares, every Shareholder present in person or by proxy, whether in a vote by a show of hands or by written ballot, shall have one vote for each Ordinary Share of record held by him. |
| 63. | A company or other corporate body being a Shareholder of the Company may duly authorize any person it deems fit to be its representative at any General Meeting or to execute or deliver a proxy on its behalf, as provided for below. Any person so authorized shall be entitled to exercise, on behalf of the corporation which he represents, all the powers which the corporation could have exercised if it were an individual Shareholder. Upon request of the chairman of the General Meeting, written evidence of such authorization (in a form reasonably acceptable to the chairman) shall be delivered to him. |
| 64. | In the case of joint holders, the vote of the senior who tenders a vote, whether in person or by proxy, shall be accepted to the exclusion of the votes of the other joint holders; and for this purpose seniority shall be determined by the order in which the names stand in the Register. |
| 65. | Shareholders may vote either personally or by proxy, or, if the Shareholder is a company or other corporate body, by a representative pursuant to Article 63 above or by a duly authorized proxy, as prescribed hereinafter. |
| 66. | Any instrument appointing a proxy or representative shall be in writing under the hand of the appointer or of his attorney duly authorized in writing, or, if such appointer is a corporation, under its common seal if any, or under the hand of some officer duly authorized in that behalf. |
| - 11 - |
| 67. | No Shareholder (or proxy or representative of a Shareholder) shall be entitled to vote at a General Meeting unless all calls or other sums presently payable in respect of his shares in the Company have been paid. |
| 68. | Every instrument of proxy, whether for a specified General Meeting or otherwise, shall be in writing in such usual or accepted form or forms as shall be approved by the Board of Directors. |
| 69. | A vote given in accordance with the terms of an instrument of appointment of proxy or representative shall be valid notwithstanding the previous death of the principal, or revocation of the appointment, or transfer of the share in respect of which the vote is given, unless notice in writing of the death, revocation or transfer shall have been received at the Office or by the chairman of the General Meeting before the vote is given. |
The Board of Directors
| 70. | (a) | The number of Directors shall be not less than three and not more than eleven, including any External Directors (as defined below). Subject to the aforesaid, the number of Directors from time to time shall be determined, from time to time, by a majority of the Directors then in office; provided that no decrease in the number of Directors shall shorten the term of any incumbent Director. |
| (b) | If at any time the Company shall be required to appoint independent or external directors, such as a public director or directors of any other type as may be required by law (“External Directors”), such directors shall serve on the Board at least in the number required by law. External Directors will be appointed, removed and serve pursuant to the relevant provisions of the law which apply to External Directors. If permitted by applicable law, External Directors will be appointed by the Board. |
| (c) | The Directors, other than External Directors required by the Companies Law (who will be chosen and appointed, will serve and whose term will expire in accordance with applicable law), shall be appointed in accordance with the provisions of this Article. |
| (d) | The Directors shall be divided into three classes, namely Class I, Class II and Class III (except for External Directors required by the Companies Law who shall not form part of any class and whose term shall be determined in accordance with applicable law and except for Directors appointed by the Board pursuant to Article 70(e) below). Initially, the Directors of each class shall be appointed or classified by Shareholder Resolution at the General Meeting at which these Articles are first adopted. Each of the classes shall be as nearly equal in number as possible. Each initial Director in Class I shall serve for a term expiring at the end of the Annual General Meeting held during the year 2015, each initial Director in Class II shall serve for a term expiring at the end of the Annual General Meeting held during the year 2016 and each initial Director in Class III shall serve for a term expiring at the end of the Annual General Meeting held during the year 2017. The initial Directors in Class I, Class II and Class III shall serve until the end of the relevant Annual General Meeting as set forth above and until their successors have been duly elected or until any such Director’s appointment terminates as provided in the Companies Law or due to any of the circumstances set forth in Article 73 below. At each Annual General Meeting, the successors to the class of Directors whose terms expire at the end of that meeting shall be elected by Shareholder Resolution to hold office for a term expiring at the end of the Annual General Meeting held in the third year following the year of their election and until their successors have been duly elected and qualified or until any such Director’s appointment terminates as provided for in the Companies Law or due to any of the circumstances set forth in Article 73 below, in such manner that after the initial terms of office set forth above, all Directors shall be appointed for terms of approximately three years, and approximately one-third of the Directors (not including External Directors) shall stand for election each year. |
| - 12 - |
| (e) | Vacancies on the Board of Directors, however arising, including as a result of an increase in the number of Directors pursuant to Article 70(a) above, may be filled by a resolution of the majority of the Directors then in office. Each Director appointed in accordance with this Article 70(e) shall hold office until the end of the next Annual General Meeting or until such Director’s appointment terminates as provided for in the Companies Law or due to any of the circumstances set forth in Article 73 below. |
| (f) | Notwithstanding the aforesaid, Directors may not be dismissed from office by the Shareholders or by a General Meeting prior to expiration of their term of office pursuant to Article 70(d) or (e) above, and the provisions of Section 230(a) of the Companies Law in this regard shall not apply. |
| (g) | Except in the case of a person nominated by the Board of Directors, no person shall be eligible to be elected as a Director unless notice in writing of the intention to nominate such person is delivered to the Office not later than ten days, and not earlier than 40 days, prior to the date scheduled for the Annual General Meeting, signed by a Shareholder entitled to participate in and vote at the scheduled meeting, together with the written consent of the proposed nominee and such information regarding the proposed nominee as would have been required to be provided to the Company and declared upon by the proposed nominee under applicable law, had such nominee been nominated, or intended to be nominated, by the board of directors of a company and any other information reasonably requested by the Company. |
| (h) | (i) | In the event the number of nominees to serve as Directors at any Annual General Meeting is greater than the number of Directors to be elected at such Annual General Meeting as determined pursuant to these Articles, the Directors elected shall be those nominees who receive the greatest number of votes up to the number of Directors to be elected. |
| (ii) | In the event the number of Directors to be elected at any Annual General Meeting (other than External Directors) is greater than the number of Directors in that class of Directors whose terms expire at such meeting, then the Annual General Meeting at which such Directors are elected shall, to the extent necessary, divide the Directors elected among the classes of Directors in order to keep the classes as nearly equal in number as possible, and the initial term of office of any additional Directors so elected to any class whose term did not expire at such meeting shall correspond to, and expire together with, the term of office of the Directors in the class to which they were elected. |
| (i) | The term of office of a Director (including an External Director) will begin as of the date of the Annual General Meeting at which he was elected or as of the date of the meeting of the Board of Directors at which he was appointed (if appointed pursuant to Article 70(e) above) or at such later date as is determined in the resolution electing or appointing him or pursuant thereto. |
| (j) | Notwithstanding any provision of these Articles, or of any law which might otherwise permit a lesser vote, and in addition to the majority required pursuant to Article 54 above with respect to amendment or replacement of these Articles, the affirmative vote of at least 75% of the voting rights of the Company represented personally or by proxy and voting thereon at a General Meeting at which a quorum is present shall be required to alter, amend or repeal this Article 70. |
| 71. | (a) | A Director shall have the right, by written notice to the Company, to appoint a person as an alternate to act in his place, to remove the alternate and appoint another in his place and to appoint an alternate in place of an alternate whose office is vacated for any reason whatsoever. A person who is not qualified to be appointed as a Director, or a person who serves as a Director or an alternate Director, may not be appointed as an alternate Director. All references in these Articles to Directors shall, where the context so requires, mean and include alternate Directors. |
| - 13 - |
| (b) | Any notice given to the Company as aforesaid shall become effective on the date fixed therein, upon delivery to the Company or, with respect to the appointment of an alternate Director, when approved by a majority of the Directors then in office, whichever is later. The approval of the appointing Director will be counted in calculating whether a majority of Directors have approved. Unless the appointing Director limits the time or scope of the appointment, the appointment is effective for all purposes until the appointing Director ceases to be a Director or terminates the appointment. |
| (c) | An alternate for a Director shall, subject to any instructions or limitations contained in the instrument appointing him, have all the authority and powers held by the Director for whom he acts as alternate; provided however, that he may not in turn appoint an alternate for himself (unless the instrument appointing him otherwise expressly provides); and provided further that an alternate shall have no standing at any Board Meeting or any meeting of a committee of the Board at which the Director appointing him is personally present or at which the Director appointing him is not entitled to participate in accordance with Article 75 below. |
| (d) | The office of an alternate for a Director shall ipso facto be vacated if he is removed by the Director appointing him, or if the office of the Director for whom he acts as alternate is vacated for any reason whatsoever, or if one of the circumstances described in Article 73 below should occur with respect to the alternate. |
| (e) | An alternate Director shall alone be responsible for his actions and omissions and shall not be deemed an agent of the Director who appointed him. |
| (f) | Every alternate Director shall be entitled to receive, so long as he serves as an alternate, notice of Board Meetings and of meetings of any relevant committees. |
| 72. | Subject to applicable law, a Director who has ceased to hold office shall be eligible for re-election or re-appointment. |
| 73. | The office of a Director shall ipso facto be vacated upon the occurrence of any of the following events: |
| (a) | His death, or, if the Director is a legal entity, it has adopted a resolution of voluntary liquidation or winding-up, or a liquidation order has been issued with respect thereto; |
| (b) | Should he be declared to be legally incompetent; |
| (c) | Should he be declared bankrupt; |
| (d) | Should he resign his office by notice in writing to the Company; or |
| (e) | As otherwise provided in the Companies Law. |
| 74. | A Director shall not be required to hold qualification shares. |
| 75. | (a) | Subject to the provisions of the Companies Law, no Director or other Office Holder of the Company shall be disqualified by his office from holding any office or place of profit within or outside the Company or with any company in which the Company shall be a shareholder or otherwise interested, or with any company which is a shareholder of, or otherwise interested in, the Company or from contracting with the Company either as vendor, purchaser or otherwise, either on his own behalf or as a director of another company or member of a firm or otherwise, nor (unless and to the extent provided otherwise in the Companies Law) shall any such contract, or any contract or arrangement entered into by or on behalf of the Company in which any Director or Office Holder shall be in any way interested, be void or voidable, nor shall he be liable to account to the Company for any profit arising from any such office or place of profit or realized by any such contract or arrangement by reason only of such Director or Office Holder holding that office or of the fiduciary relations thereby established, but it is hereby declared that the nature of his interest must be disclosed by him as provided in the Companies Law and in any event not later than at the Board Meeting at which the contract or arrangement is first taken into consideration, if his interest then exists or, in any other case, at the first Board Meeting after the acquisition of his interest. |
| - 14 - |
| (b) | Unless and to the extent provided otherwise in the Companies Law, every Director shall be entitled, after such disclosure, to vote as a Director in respect of any contract or arrangement in which he is so interested as aforesaid. Unless and to the extent provided otherwise in the Companies Law, a general notice that a Director is a member of any firm or company and is to be regarded as interested in all transactions with that firm or company shall be a sufficient disclosure under this Article as regards such Director and the said transactions, and after such general notice (unless and to the extent provided otherwise in the Companies Law), it shall not be necessary for such Director to give a special notice relating to any particular transaction with that firm or company. |
| (c) | A transaction referred to in this Article 75, which is not an Extraordinary Transaction shall be approved by the Board or by a committee authorized to do so by the Board. Such approval may be general in nature and may be given in advance. Notwithstanding the aforesaid, if according to the provisions of the Companies Law a specific or special approval for a particular transaction or type of transaction is required, such transaction shall also require such approval. |
| (d) | An Extraordinary Transaction requires approval as provided under the Companies Law. |
| 76. | (a) | A Director may be paid remuneration by the Company for his services as a Director to the extent such remuneration is approved pursuant to the Companies Law. |
| (b) | If a Director, willing to do so, is called upon to fulfill special services or make special efforts for any of the Company’s objects, by travelling abroad or staying there or otherwise, the Company may pay him a salary at a fixed rate or a percentage of its profits or otherwise as the Board of Directors may decide and subject to approval by Shareholder Resolution and the provisions of the Companies Law, and such salary may be in addition to or in place of the fixed remuneration (if any). |
Proceedings of the Board of Directors
| 77. | (a) | The Chairman of the Board of Directors shall convene Board Meetings in accordance with the provisions of the Companies Law, and may adjourn and otherwise regulate the proceedings of such meetings, as he thinks fit. The quorum for Board Meetings and/or for any matter to be brought before the Board shall be a majority of the Directors then in office and entitled to participate and vote with respect thereto. |
| (b) | Unless and to the extent provided otherwise in the Companies Law, a Director who is an interested party in any transaction shall be counted for purposes of a quorum despite his interest. |
| (c) | A Director may participate personally or by his alternate. |
| 78. | Notice of a Board Meeting may be given verbally, by telephone or sent to all Directors at their registered addresses, by telex, facsimile, electronic mail or other reliable method of transmission, at least 24 hours prior to the Board Meeting unless all Directors agree to shorter notice. Directors will be entitled to participate by way of video or audio conference in such manner that all persons participating in the meeting are able to hear each other at the same time, and the Company will cooperate, as may reasonably be required, in providing video or audio conferencing capabilities to effectuate such participation. |
| 79. | (a) | Each Director shall have one vote. |
| - 15 - |
| (b) | All resolutions of the Board will be adopted by a simple majority of the Directors present and voting (with the Directors participating by video or audio conference, if any, being deemed present and entitled to vote) at a Board Meeting. |
| 80. | The Board of Directors shall elect one of its members to be the Chairman of the Board of Directors, and may remove such Chairman from office and appoint another in his place. The Chairman of the Board of Directors shall take the chair at every Board Meeting, but if there is no such Chairman, or if he shall have indicated in advance that he will not be attending, or if at any meeting he is not present within 15 minutes of the time appointed for the meeting, or if he is unwilling to take the chair, the Directors present shall choose one of their number to be the Chairman of such meeting. |
| 81. | The Chairman of a Board Meeting, whether he be the Chairman of the Board of Directors or any other member of the Board of Directors, shall have no extra or casting vote. |
| 82. | A Board Meeting at which a quorum is present shall be competent to exercise all the authorities, powers and discretions for the time being vested in or exercisable by the Board of Directors. |
| 83. | (a) | Subject to applicable law, the Board of Directors may for any particular matter delegate any or all of its powers to committees consisting of one or several Directors, as the Board of Directors may deem fit, including, as aforesaid, the authority to approve transactions that are not Extraordinary Transactions, pursuant to Sections 270(l) and 271 of the Companies Law, and the Board of Directors may from time to time revoke such delegation. |
| (b) | Any committee so formed shall, in the exercise of the powers so delegated, conform to any regulations that may be imposed on it by the Board of Directors. The meetings and proceedings of any such committee consisting of two or more members shall be governed by the provisions herein contained for regulating the meetings of the Board of Directors, so far as the same are applicable thereto, and so far as not superseded by any regulations made by the Board of Directors under this Article. |
| 84. | All acts performed at or in accordance with any Board Meeting, or any meeting of a committee of the Board of Directors, or by any person acting as Director or alternate for a Director, shall, notwithstanding that it may afterwards be discovered that there was some defect in the appointment of such Directors or members of a committee of the Board of Directors or person acting as aforesaid or any of them, or that they or any of them were disqualified, be as valid as if every such person had been duly appointed and was qualified to be a Director, alternate or a member of such a committee, as the case may be. |
| 85. | A resolution in writing signed by all Directors or members of a committee of the Board of Directors then in office and entitled to vote thereon or to which all such Directors or members shall have given their written consent (by letter, telegram, facsimile, electronic mail or otherwise) shall be deemed to have been unanimously adopted by a Board Meeting or committee meeting duly convened and held. |
Managing Directors or General Manager
| 86. | The Board of Directors may from time to time appoint one or more persons (whether a Director or not) to be managing director(s), general manager(s), chief executive officer(s) and/or president(s) (or any similar function with a different title) of the Company, either for a fixed term or without any limitation as to the period for which he is or they are to hold office, and may from time to time modify or revoke such titles or (subject to any provisions of any contract between him or them and the Company) remove or dismiss him or them from office and appoint another or others in his or their place or places. |
| 87. | Subject to the provisions of the Companies Law, the remuneration of a managing director, general manager, chief executive officer and/or president shall from time to time (subject to any contract between him and the Company) be fixed by the Board of Directors, and may be in the form of a fixed salary or commission on dividend, profits or turnover of the Company, or of any other company the Company has an interest in, or by participation in profits or in one or more of these forms. |
| - 16 - |
| 88. | Subject to the provisions of the Companies Law, the Board of Directors may from time to time entrust to and confer upon a managing director, general manager, chief executive officer and/or president for the time being such of the powers exercisable under these Articles by the Board of Directors as it may think fit, and may confer such powers for such time, and to be exercised for such objects and purposes, and upon such terms and conditions, and with such restrictions, as it thinks expedient; and it may confer such powers, either collaterally with, or to the exclusion of, and in substitution for, all or any of the powers of the Board of Directors in that behalf; and may from time to time revoke, withdraw, alter or vary all or any of such powers. |
Powers of the Board of Directors
| 89. | The management of the business of the Company shall be vested in the Board of Directors, and the Board of Directors may exercise all such powers and do all such acts and things as the Company is, by its Articles of Association or under the law, authorized to exercise and do, and are not hereby or by statute directed or required to be exercised or done by the Company in General Meeting, but subject, nevertheless, to the provisions of the Companies Law and to these Articles. |
| 90. | Without prejudice to any of the general powers granted to the Board of Directors in accordance with Article 89 above and any other powers granted to it under these Articles, and without restricting or reducing in any way any of the above mentioned powers, it is hereby explicitly declared that the Board of Directors shall have the following powers: |
| (a) | To appoint a person or persons (whether they be incorporated or not) to receive and hold in trust for the Company any property whatsoever that belongs to the Company or that the Company has an interest in, or for any other purpose and to execute and perform all actions, deeds and necessary activities with relation to any such trust, and to see to the remuneration of any such trustee(s). |
| (b) | To initiate, manage, defend, compromise or discontinue any and all legal proceedings on behalf of or against the Company or its officials or that pertain in any way to its affairs, and to compromise and extend the period for payment or discharge of any debt due or suits or claims by or against the Company. |
| (c) | To refer any suit or claim by or against the Company to arbitration. |
| (d) | To determine, from time to time, those authorized to sign in the Company’s name on bills of exchange, promissory notes, receipts, certificates of receipt, endorsements, checks, certificates of dividend, releases, contracts and other documents of any kind whatsoever. |
| (e) | In general, and subject to the provisions of the Companies Law and these Articles, to delegate to any person, firm, company or variable group of people, the powers, authority and discretion vested in the Board of Directors. |
Local Management
| 91. | The Board of Directors may from time to time provide for the management and transaction of the affairs of the Company in any specified locality, whether in Israel or abroad, in such manner as it thinks fit, and the provisions contained in the next following Article shall be without prejudice to the general powers conferred by this Article on the Board of Directors. |
| - 17 - |
| 92. | The Board of Directors may from time to time, and at any time, establish any local board or agency for managing any of the affairs of the Company in any specified locality, in Israel or abroad, and may appoint any person to be a member of such local board, or any manager or agent, and may fix their remuneration. Subject to the provisions of the Companies Law, the Board of Directors may from time to time, and at any time, delegate to any person so appointed any of the powers, authority and discretions for the time being vested in the Board of Directors, and may authorize any member for the time being of any such local board to continue in his office notwithstanding any vacancy which may occur, and any such appointment or delegation may be made on such terms and subject to such conditions as the Board of Directors may think fit, and the Board of Directors may at any time remove any person so appointed and may annul or vary any such delegation. The Board of Directors may authorize any person to whom it has delegated powers, authority or discretion, as mentioned, to delegate them or part of them further. |
Register of Shareholders
| 93. | (a) | The Company shall keep a Register in which it may record such information as may be deemed appropriate by the Board of Directors and/or as may be permitted by the Companies Law or these Articles. In addition, the Company shall record in the Register the following information: |
| (i) | The names and addresses of the Shareholders, the number of shares held by each Shareholder and the amount paid or the amount to be considered as paid on the shares of each Shareholder; |
| (ii) | The day each person was registered in the Register as a Shareholder; |
| (iii) | The amounts called, if any, that are due on the shares of each Shareholder; and |
| (iv) | Any other information required by the Companies Law or these Articles to be recorded in the Register. |
| (b) | The principal register shall be kept at the Office and, apart from the times the Register is closed in accordance with the provisions of the Companies Law or these Articles, shall be open to the inspection of any Shareholder free of charge, and of any other person at such fee as the Company shall determine for each matter, during regular business hours. |
| (c) | The Register may be closed for such period, if any, as the Board of Directors shall determine from time to time, on the condition that the Register shall not be closed for a period exceeding 30 days during any calendar year. |
Minutes and the Seal
| 94. | (a) | The Board of Directors shall cause minutes to be duly recorded regarding the names of the Directors present at each Board Meeting and each meeting of any committee(s) of the Board of Directors; the names of the Shareholders present at each General Meeting; and the proceedings and resolutions of General Meetings and of Board Meetings and meetings of committee(s) of the Board of Directors. Any minutes as aforesaid of a Board Meeting, of a meeting of a committee of the Board of Directors or of a General Meeting, if purporting to be signed by the chairman of such meeting or by the chairman of the next succeeding meeting, shall be accepted as prima facie evidence of the matters therein recorded. |
| (b) | (i) | The Company may have one or more rubber stamps for affixing on documents. |
| (ii) | The Board of Directors shall be entitled to authorize any person or persons (even if he or they is or are not Directors(s) of the Company) to act and sign on behalf of the Company, and further to delegate such signatory powers, and the acts and signatures of such person or persons on behalf of the Company shall bind the Company insofar as such person or persons acted and signed within his or their powers aforesaid. |
| (iii) | The Board of Directors may provide for a seal. If the Board of Directors so provides, it shall also provide for the safe custody thereof; such seal shall not be used except by the authority of the Board of Directors. |
| - 18 - |
The Secretary, Officers and Attorneys
| 95. | The Board of Directors may appoint a corporate secretary to the Company and may appoint officers, personnel, agents and servants, for fixed, provisional or special duties, as the Board of Directors may from time to time deem fit, and may from time to time, in its absolute discretion, suspend the service of any one or more of such persons. |
| 96. | The Board of Directors may determine the powers and duties, as well as the salaries, of such persons and may demand security in such cases and in such amounts as it deems fit. |
| 97. | The Board of Directors may from time to time, and at any time, by power of attorney, appoint any company, firm or person or body of persons, whether nominated directly or indirectly by the Board of Directors, to be the attorney(s) of the Company for such purposes and with such powers, authorities and discretions (not exceeding those vested in or exercisable by the Board of Directors under these Articles), and for such period and subject to such conditions as it thinks fit, and any such power of attorney may contain such provisions for the protection and convenience of the above-mentioned attorney(s) and/or of those persons who come into contact with such attorney(s) as the Board of Directors may think fit, and may also authorize any such attorney(s) to delegate all or any of the powers, authorities and discretion vested in him or them. |
Dividends and Reserve Fund
| 98. | The Board of Directors may, from time to time, set aside, out of the profits of the Company, such sums as it thinks proper, as a reserve fund to meet contingencies, or for equalizing dividends, or for special dividends, or for repairing, improving and maintaining any of the property of the Company, and for such other purposes as the Board of Directors shall in its absolute discretion think conducive to the interests of the Company, and may invest the sums so set aside in such investments as it may think fit, and from time to time deal with and vary such investments, and dispose of all or any part thereof for the benefit of the Company, and may divide the reserve fund into such special funds as it thinks fit, and employ the reserve fund or any part thereof in the business of the Company, and that without being bound to keep the same separate from the other assets of the Company. The Board of Directors may also, without placing the same to reserve, carry forward any profits which it deems prudent not to divide. |
| 99. | Subject to the rights of holders of shares with limited or preferred rights as to dividends, and subject to the provisions of these Articles as to the reserve fund, all dividends shall be paid to the Shareholders in proportion to the amount paid up or credited as paid up on account of the nominal value of the shares held by them respectively and in respect of which such dividend is being paid, without regard to any premium paid in excess of the nominal value, if any, but if any share is issued on terms providing that it shall rank for dividend from a particular date, such share will rank for dividend accordingly. |
| 100. | Subject to the provisions of the Companies Law, the Board of Directors may from time to time declare such dividends as may appear to the Board of Directors to be justified by the profits of the Company, and cause the Company to pay such dividends. The Board of Directors shall have the full authority to determine the time for payment of such dividends, and the record date for determining the Shareholders entitled thereto, provided such date is not prior to the date of the resolution to distribute the dividend, and no Shareholder who shall be registered in the Register with respect to any shares after the record date so determined shall be entitled to share in any such dividend with respect to such shares. |
| 101. | No dividend shall be paid other than out of the profits of the Company, as defined in the Companies Law, and no interest shall be paid by the Company on dividends. |
| - 19 - |
| 102. | A dividend may be paid, wholly or partly, by the distribution of specific assets of the Company or by the distribution of specific assets, paid-up shares, debentures or debenture stock of any other company, or in any one or more such ways. |
| 103. | The Board of Directors may resolve that any moneys, investments or other assets forming part of the undivided profits of the Company standing to the credit of the reserve fund, or to the credit of any reserve fund for the redemption of capital, or to the credit of any reserve fund for the revaluation of real estate or other assets of the Company or any other reserve fund or investment funds, or in the hands of the Company and available for dividends, or representing premiums received on the issue of shares and standing to the credit of the share premium account, be capitalized and distributed among such of the Shareholders as would be entitled to receive the same if distributed by way of dividend and in the same proportion on the basis that they become entitled thereto as capital; and that all or any part of such capitalized fund be applied on behalf of such Shareholders in paying up in full, either at par or at such premiums as the resolution may provide, any unissued shares or debentures or debenture stock of the Company which shall be distributed accordingly or in or towards the payment, in full or in part, of the uncalled liability on any issued shares or debentures or debenture stock; and that such distribution or payment shall be accepted by such Shareholders in full satisfaction of their share and interest in the said capitalized sum. |
| 104. | For the purpose of giving effect to any resolution under the two last preceding Articles, the Board of Directors may settle any difficulty which may arise in regard to the distribution as it thinks expedient, and, in particular, without derogating from the generality of the foregoing, may issue fractional Share Certificates or make payment in lieu of fractional shares in an amount determined by the Board, and may fix the value for distribution of any specific assets, and may determine that cash payments shall be made to any Shareholders upon the basis of the value so fixed, or that fractions of less than NIS 0.01 (one New Agora) in value may be disregarded in order to adjust the rights of all parties, and may vest any such cash, shares, debentures, debenture stock or specific assets in trustees for the persons entitled to the dividend or capitalized fund against such securities as may seem expedient to the Board of Directors. Where requisite, a proper contract shall be filed in accordance with the Companies Law, and the Board of Directors may appoint any person to sign such contract on behalf of such persons entitled to the dividend or capitalized fund. |
| 105. | The Board of Directors may deduct from any dividend, bonus or other amount to be paid in respect of shares held by any Shareholder, whether alone or together with another Shareholder, any sum or sums due from him and payable by him alone or together with any other person to the Company on account of calls or the like. |
| 106. | (a) | The Board of Directors may retain any dividend or other monies payable or property distributable in respect of a share on which the Company has a lien, and may apply the same in or towards satisfaction of the debts, liabilities, or engagements in respect of which the lien exists. |
| (b) | The Board of Directors may, when paying any dividend, resolve to retain any dividend, or other monies payable or property distributable, for distribution with respect to a share in respect of which any person is under these Articles entitled to become a Shareholder, or which any person is under these Articles entitled to transfer, until such person shall become a Shareholder in respect of such share or shall transfer the same. |
| 107. | All unclaimed dividends or other monies payable in respect of a share may be invested or otherwise made use of by the Board of Directors for the benefit of the Company until claimed. The payment by the Board of Directors of any unclaimed dividend or such other monies into a separate account shall not constitute the Company a trustee in respect thereof. The principal (and only the principal) of an unclaimed dividend or such other moneys shall, if claimed, be paid to a person entitled thereto. Any dividend unclaimed after a period of three (3) years from the date of declaration of such dividend shall be forfeited to the benefit of the Company; provided, however, that the Company, at its sole discretion, shall be entitled (but not required) to pay any such dividend, or any part thereof, as provided above, to a person who would have been entitled thereto had the same not been forfeited. |
| - 20 - |
| 108. | Any dividend or other monies payable in cash in respect of a share may be paid by check or warrant sent through the post to, or left at, the registered address of the person entitled thereto or by transfer to a bank account specified by such person (or, if two or more persons are registered as joint holders of such share to the one whose name appears first in the Register), or to such person and at such address as the person entitled thereto may by writing direct. Every such check or warrant shall be made payable to the order of the person to whom it is sent, or to such person as the person entitled thereto as aforesaid may direct, and payment of the check or warrant by the banker upon whom it is drawn shall be a good discharge to the Company. |
| 109. | If several persons are registered as joint holders of any share, or are entitled jointly thereto in consequence of the death or bankruptcy of the holder or otherwise, any one of them may give effectual receipts for any dividend payable or property distributable on the share. |
Books of Account
| 110. | The Board of Directors shall cause accurate books of account to be kept in accordance with the provisions of the Companies Law and any other applicable law. The books of account shall be kept at the Office or at any other place or places as the Board of Directors may deem fit, and they shall always be open to inspection by Directors. No Shareholder not being a Director shall have the right to inspect any account or book or document of the Company except as conferred by law or authorized by the Board of Directors. |
Accounts and Audit
| 111. | Once at least in every year, the accounts of the Company shall be examined and the correctness of the profit and loss account and balance sheet ascertained by a duly qualified auditor. |
| 112. | The appointment, authorities, rights, salaries and duties of the auditor or auditors shall be regulated by the law in force for the time being and by the provisions of these Articles; provided, however, that the Board of Directors shall fix the remuneration of the auditor(s). |
Notices
| 113. | Without derogating from Article 52 above or Article 122 below, any notice or document may be served by the Company upon any Shareholder either personally or by sending it by prepaid mail (air mail if sent from Israel to a place outside Israel) addressed to such Shareholder at his address as described in the Register or such other address (if any) as he may have designated in writing for the receipt of notices and documents. Any notice or document may be served by any Shareholder upon the Company by tendering the same in person to the managing director/general manager/chief executive officer/president of the Company at the Office or by sending it by prepaid registered mail (air mail if posted outside Israel) to the Company at the Office. Any such notice or document shall be deemed to have been served 48 hours after it has been posted (seven days if sent from Israel to a place outside Israel, or if sent to Israel from a place outside Israel), or when actually received by the addressee if sooner than 48 hours or seven days, as the case may be, after it has been posted, or when actually tendered in person, to such Shareholder (or to the managing director/general manager/chief executive officer/president); provided, however, that notice may be sent by cablegram, electronic mail, telex, facsimile or other customary method and confirmed by mail as aforesaid, and such notice shall be deemed to have been given the first business day after such cablegram, electronic mail, telex, facsimile or other customary method has been sent or when actually received by such Shareholder (or by the Company), whichever is earlier. If a notice is, in fact, received by the addressee, it shall be deemed to have been duly served when received, notwithstanding that it was defectively addressed, or failed in some other respect, to comply with the provisions of this Article. |
| 114. | A notice may be given by the Company to the joint holders of a share by giving notice to the joint holder named first in the Register in respect of the share. |
| 115. | Without derogating from Article 52 above, any Shareholder whose address is not described in the Register, and who shall not have designated in writing an address for the receipt of notices, shall not be entitled to receive any notice from the Company. |
| - 21 - |
| 116. | The Company may declare that any document(s) will be delivered or be available for review at the Office or any other place designated by the Board of Directors. |
| 117. | Whenever it is required to give prior notice or publicize a specified number of days in advance or where a notice or publication is valid for a specified period, the day of the publication or the day of service of the notice shall be included in such count or period. |
| 118. | Service of notice to a relative of a Shareholder living at the same address with him will be deemed service to such Shareholder. |
| 119. | Subject to applicable law, any Shareholder, Director or other person entitled to receive notice in accordance with these Articles or law may waive notice, in advance or retroactively, in a particular case or type of case or generally, and if so, notice will be deemed as having been duly served, and all proceedings or actions for which the notice was required will be deemed valid. |
| 120. | Any person entitled to a share by operation of law or by transfer, transmission or otherwise will be bound by any notice served or by any publication made pursuant to these Articles with respect to such share prior to his being registered in the Register as owner of the shares. |
| 121. | It shall not be necessary to set forth in detail in any publication as provided for in Article 52(b) above, the full text of any proposed resolutions and a general description of the nature of the matters on the agenda will suffice. The Company shall be entitled, however, but shall be under no obligation to do so, to specify in any publication in respect of a meeting, a place and a time where and when the full text of proposed resolution(s) may be reviewed. |
| 122. | Notwithstanding anything to the contrary contained herein, the Company may give notice to any Shareholder by filing an appropriate periodic report with the SEC, by posting a notice on the Company’s website, by publishing in one or more international wire services or in one or more newspapers or by publicizing in any other manner reasonably determined by the Company, and the date of such filing, posting or other publication shall be deemed the date on which such notice has been served upon such Shareholders. Where notice is given by more than one method, it will be deemed served on the earliest of such dates. |
| 123. | The accidental omission to give notice to any Shareholder pursuant to any applicable law or these Articles or the non-receipt of any such notice by any Shareholder entitled to receive notice shall not invalidate any action, transaction, resolution or proceedings taken by the Company and/or at or by any General Meeting. |
Winding-Up
| 124. | If the Company shall be wound up, then, subject to applicable law and the rights of holders of shares with limited or preferred rights, the assets of the Company available for distribution among the Shareholders shall be distributed to them in proportion to the amount paid up or credited as paid up on account of the nominal value of the shares held by them respectively and in respect of which such distribution is being made, without regard to any premium paid in excess of the nominal value, if any. |
Indemnification, Insurance and Exemption
| 125. | (a) | The Company may, subject and pursuant to the provisions of the Companies Law, indemnify an Office Holder of the Company for all liabilities and expenses incurred by him arising from or as a result of any act (or omission) carried out by him as an Office Holder of the Company and which is indemnifiable pursuant to applicable law, to the fullest extent permitted by law, including with respect to the following: |
| (i) | Monetary liabilities or obligations imposed on the Office Holder in favor of another person pursuant to a court judgment, including a compromise judgment or an arbitrator’s decision approved by a court; |
| - 22 - |
| (ii) | Payments which the Office Holder is obligated to make to an injured party as set forth in Section 52(54)(a)(1)(a) of the Securities Law and expenses the Office Holder incurred in connection with a proceeding under Chapters H’3, H’4 or I’1 of the Securities Law, including reasonable litigation expenses, including attorney’s fees, or in connection with Article D of Chapter Four of Part Nine of the Companies Law; |
| (iii) | Reasonable litigation expenses, including attorney’s fees, incurred by the Office Holder in consequence of an investigation or proceeding conducted against the Office Holder by an authority that is authorized to conduct such investigation or proceeding, and which was concluded without the submission of an indictment against the Office Holder and without imposing on the Office Holder any financial obligation in lieu of criminal proceedings, or which was concluded without the submission of an indictment against the Office Holder but with imposing on such Office Holder a financial obligation in lieu of criminal proceedings in respect of an offense that does not require proof of criminal intent or in connection with a financial sanction; |
For the purposes hereof: (i) “a proceeding concluded without the submission of an indictment in a matter in respect of which a criminal investigation was conducted”; and (ii) “financial obligation in lieu of a criminal proceeding”, shall have the meanings specified in Section 260(a)(1A) of the Companies Law;
| (iv) | Reasonable litigation expenses, including attorney’s fees incurred by the Office Holder or imposed upon him by a court, in a proceeding brought against the Office Holder by the Company or on its behalf or by another person, or in a criminal action in which the Office Holder is acquitted, or in a criminal action in which the Office Holder is convicted of an offense that does not require proof of criminal intent; |
| (v) | Expenses incurred by the Office Holder in connection with a proceeding under Chapter G’1, of the Restrictive Trade Law, including reasonable litigation expenses, including attorney’s fees; |
| (vi) | Any other liability, obligation or expense indemnifiable or which may from time to time be indemnifiable by law. |
The Company may indemnify an Office Holder post-factum and may also undertake in advance to indemnify an Office Holder, provided that: (x) an undertaking in advance to indemnify an Office Holder with respect to the matters specified in Article 125(a)(i) above is limited to types of occurrences which, in the opinion of the Board of Directors, in light of the Company’s actual activities at the time of the undertaking, are foreseeable and to an amount or to criteria the Board of Directors has determined to be reasonable in the circumstances; and (y) in the undertaking in advance to indemnify an Office Holder, the types of occurrences that the Board of Directors believes to be foreseeable in light of the Company’s actual activities at the time the undertaking to indemnify was given are mentioned, as is the amount or criteria that the Board of Directors determined to be reasonable in the circumstances.
| (b) | The Company may, subject and pursuant to the provisions of the Companies Law, enter into contracts to insure the liability of Office Holders of the Company for any liabilities or expenses incurred by or imposed upon them arising from or as a result of any act (or omission) carried out by them as Office Holders of the Company, to the fullest extent permitted by law, including in respect of any liability imposed on any Office Holder with respect to any of the following: |
| (1) | A breach of his duty of care to the Company or to any other person; |
| - 23 - |
| (2) | A breach of his duty of loyalty to the Company, provided that the Office Holder acted in good faith and had a reasonable basis to believe that such act would not prejudice the interests of the Company; |
| (3) | Monetary liabilities or obligations imposed on him in favor of another person; |
| (4) | A payment which the Office Holder is obligated to make to an injured party as set forth in Section 52(54)(a)(1)(a) of the Securities Law and expenses that the Office Holder incurred in connection with a proceeding under Chapters H’3, H’4 or I’1 of the Securities Law, including reasonable litigation expenses, including attorney’s fees, or in connection with Article D of Chapter Four of Part Nine of the Companies Law; |
| (5) | Expenses incurred by the Office Holder in connection with a proceeding under Chapter G’1, of the Restrictive Trade Law, including reasonable litigation expenses, including attorney’s fees. |
| (c) | The Company may, to the fullest extent permitted by law, exempt and release an Office Holder of the Company, including in advance, from and against all or part of his liability for monetary or other damages due to, or arising or resulting from, a breach of his duty of care to the Company. The Directors of the Company are released and exempt from all liability as aforesaid to the fullest extent permitted by law with respect to any such breach, which has been or may be committed. |
| (d) | The Company may, subject to the provisions of the Companies Law, procure insurance for, indemnify and/or exempt and release any person who is not an Office Holder including, without limitation, any employee, agent, consultant or contractor of the Company who is not an Office Holder. |
| (e) | The Company may, as aforesaid, indemnify, insure and exempt from liability any Office Holder to the fullest extent permitted by applicable law. Accordingly: (i) any amendment to the Companies Law, the Securities Law, the Restrictive Trade Law or any other applicable law expanding the ability of the Company to indemnify, insure or exempt from liability any Office Holder, or expanding the right of any Office Holder to be indemnified, insured or exempted from liability, beyond or in addition to the provisions of these Articles, shall, to the fullest extent possible, automatically and immediately apply to the Office Holders of the Company and be deemed as included in these Articles to the fullest extent permitted by applicable law; and (ii) any amendment to the Companies Law, the Securities Law, the Restrictive Trade Law or any other applicable law adversely affecting the ability of the Company to indemnify, insure or exempt from liability any Office Holder or adversely affecting the right of any Office Holder to be indemnified, insured or exempted from liability as provided for in these Articles shall have no effect post factum and shall not affect the Company’s obligations or ability to indemnify, insure or exempt from liability an Office Holder for any act (or omission) carried out prior to such amendment, unless otherwise provided by applicable law. |
Forum for Adjudication of Disputes
| 126. | (a) | Unless the Company consents in writing to the selection of an alternative forum, with respect to any causes of action arising under the U.S. Securities Act of 1933 as amended, against any person or entity, including such claims brought against the Company, its directors, officers, employees, advisors, attorneys, accountants or underwriters (who, in each case, shall be deemed third party beneficiaries of this Article 126), the federal district courts of the United States of America shall be the exclusive forum for the resolution of any complaint asserting a cause of action arising under the U.S. Securities Act of 1933, as amended; and |
| (b) | unless the Company consents in writing to the selection of an alternative forum, the competent courts in Tel Aviv, Israel shall be the exclusive forum for (i) any derivative action or proceeding brought on behalf of the Company, (ii) any action asserting a claim of breach of a fiduciary duty owed by any director, officer or other employee of the Company to the Company or the Company’s shareholders, or (iii) any action asserting a claim arising pursuant to any provision of the Companies Law or the Securities Law. Any person or entity purchasing or otherwise acquiring or holding any interest in shares of the Company shall be deemed to have notice of and consented to these provisions. This Article 126 shall not apply to causes of action arising under the U.S. Exchange Act of 1934, as amended. |
Exhibit 4.7
AMENDMENT TO PERSONAL EMPLOYMENT AGREEMENT
THIS AMENDMENT (the “Amendment”) to the Employment Agreement dated December 23, 2013 (the “Employment Agreement”) is entered into this 28 day of December, 2021 (the “Effective Date”), by and between Galmed Research and Development Ltd., having its place of business at 16 Tiomkin Street, Tel Aviv, 6578317, Israel (the “Company”), and Allen Baharaff, I.D. No 059100818, residing at 7 Hayotman St. Tel Aviv, Israel, Israel (the “Executive”).
| WHEREAS, | the Company and the Executive are parties to the Employment Agreement; and |
| WHEREAS, | the board of directors of the Company have resolved to approve certain amendment to the Employment Agreement, subject to the approval of the shareholders which was obtained on August 30, 2021; |
NOW, THEREFORE, in consideration of the premises and mutual agreement hereinafter contained, the parties agree as follows:
| 1. | Except as provided explicitly herein, all other provisions of the Employment Agreement (including any of its Exhibits and Amendments) shall continue in full force and effect, mutatis mutandis. All capitalized terms used in this Amendment and not defined herein shall have the meanings ascribed to then in the Employment Agreement. |
| 2. | As of January 1, 2021, Section 6 of Annex A of the Employment Agreement shall be replaced in its entirely by the following: |
“NIS 170,000”
As of January 1, 2021, any references in the Employment Agreement to the term “Monthly Salary” shall refer to the amount set forth in this Section.
| 3. | Section 9(c) of Annex A of the Employment Agreement shall be replaced in its entirely by the following: |
“Unless otherwise agreed by the Board and subject to applicable law, vacation days may be accumulated for no more than two years. Unused accumulated vacation days that exceed the number of vacation days that may be accumulated over a two-year period (currently, 48 vacation days) shall be redeemed once a year, on March 1st, provided that the redemption will not result in the number of accumulated vacation days following the redemption being less than 48 days, or as otherwise required by law. Accumulated vacation days shall also be redeemed in the event of termination of employment.”
| 4. | This Amendment supersedes all prior agreement, written or oral, between the Parties relating to the subject matter of this Amendment. This Amendment may be executed in two or more counterparts, each of which shall constitute an original and all of which shall be deemed a single agreement. Except as amended hereby, the Employment Agreement remains in effect and unmodified. In the event of any conflict or inconsistency between the provisions of the Employment Agreement and this Amendment, the provisions of this Amendment shall prevail. |
IN WITNESS WHEREOF, the parties have executed this Amendment as of the date first above written.
GALMED RESEARCH AND DEVELOPMENT LTD. |
Executive | |||
| Name: | Amir Poshinsky | Name: | Allen Baharaff | |
| Title: | Chairman of the Audit Committee | Title: | President and CEO | |
| Date | December 28, 2021 | Date: | December 28, 2021 | |
Exhibit 12.1
CERTIFICATION
I, Allen Baharaff, certify that:
1. I have reviewed this annual report on Form 20-F of Galmed Pharmaceuticals Ltd. (the “Company”);
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the Company as of, and for, the periods presented in this report;
4. The Company’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the Company and have:
(a) Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the Company, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
(b) Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
(c) Evaluated the effectiveness of the Company’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
(d) Disclosed in this report any change in the Company’s internal control over financial reporting that occurred during the period covered by the annual report that has materially affected, or is reasonably likely to materially affect, the Company’s internal control over financial reporting; and
5. The Company’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the Company’s auditors and the audit committee of the Company’s board of directors (or persons performing the equivalent functions):
(a) All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the Company’s ability to record, process, summarize and report financial information; and
(b) Any fraud, whether or not material, that involves management or other employees who have a significant role in the Company’s internal control over financial reporting.
| By: | /s/ Allen Baharaff | |
| Allen Baharaff | ||
| President and Chief Executive Officer |
Date: May 2, 2022
Exhibit 12.2
CERTIFICATION
I, Yohai Stenzler, certify that:
1. I have reviewed this annual report on Form 20-F of Galmed Pharmaceuticals Ltd. (the “Company”);
2. Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
3. Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the Company as of, and for, the periods presented in this report;
4. The Company’s other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the Company and have:
(a) Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the Company, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
(b) Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
(c) Evaluated the effectiveness of the Company’s disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
(d) Disclosed in this report any change in the Company’s internal control over financial reporting that occurred during the period covered by the annual report that has materially affected, or is reasonably likely to materially affect, the Company’s internal control over financial reporting; and
5. The Company’s other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the Company’s auditors and the audit committee of the Company’s board of directors (or persons performing the equivalent functions):
(a) All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the Company’s ability to record, process, summarize and report financial information; and
(b) Any fraud, whether or not material, that involves management or other employees who have a significant role in the Company’s internal control over financial reporting.
| By: | /s/ Yohai Stenzler | |
| Yohai Stenzler | ||
| Chief Accounting Officer |
Date: May 2, 2022
Exhibit 13.1
CERTIFICATION PURSUANT TO
18 U.S.C. SECTION 1350,
AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the annual report of Galmed Pharmaceuticals Ltd. (the “Company”) on Form 20-F for the period ending December 31, 2021, as filed with the Securities and Exchange Commission on the date hereof (the “Report”), the undersigned hereby certify that to the best of our knowledge:
| (1) | The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and | |
| (2) | The information contained in the Report fairly presents, in all material respects, the financial condition and results of operation of the Company. |
| By: | /s/ Allen Baharaff | |
| Allen Baharaff | ||
| President and Chief Executive Officer |
| By: | /s/ Yohai Stenzler | |
| Yohai Stenzler | ||
| Chief Accounting Officer |
Date: May 2, 2022
The certification set forth above is being furnished as an exhibit solely pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 and is not being filed as part of the annual report on Form 20-F for the period ended December 31, 2021, or as a separate disclosure document of the Company or the certifying officers.
Exhibit 15.1
CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
We consent to the incorporation by reference in the Registration Statements on Form S-8 (Registration No. 333-206292 and 333-227441) and the Company’s Registration Statement on Form F-3 (Registration No. 333-254766) of our report dated May 2, 2022 relating to the consolidated financial statements of Galmed Pharmaceuticals Ltd., (the “Company”), which appear in the Company’s Annual Report on Form 20-F for the year ended December 31, 2021.
| By: | /s/ Brightman Almagor Zohar & Co. | |
Brightman Almagor Zohar & Co. Certified Public Accountants | ||
| Date: May 2, 2022 | A Firm in the Deloitte Global Network |