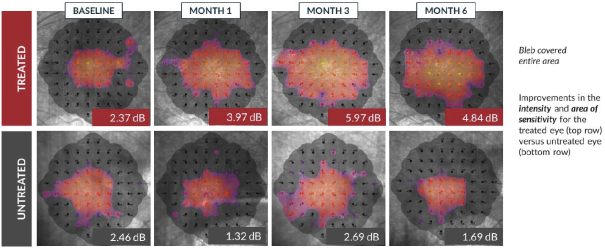
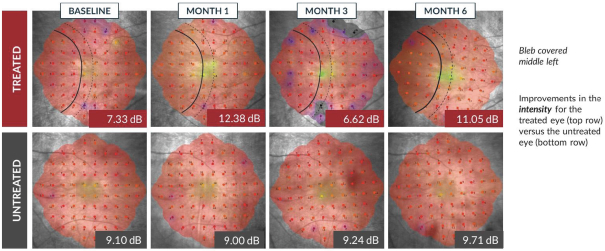
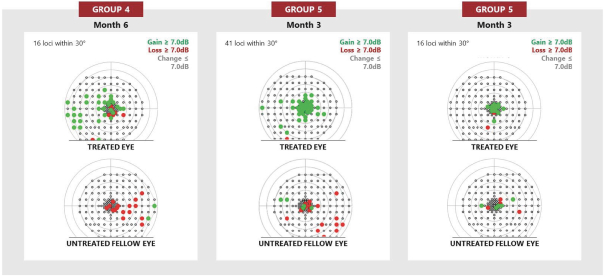
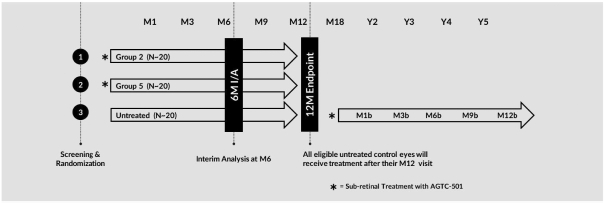
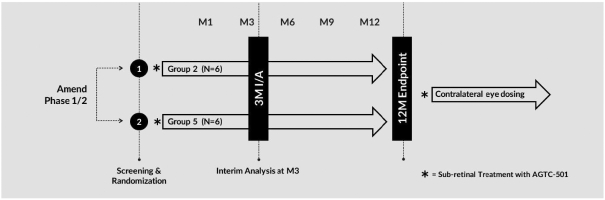
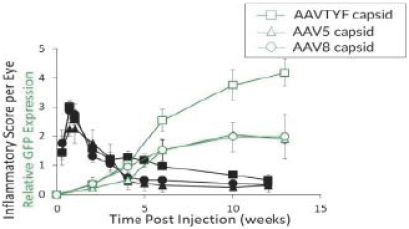
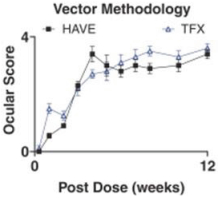
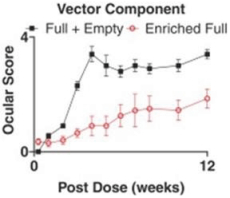
and insufficient purity. Our focus is to develop an integrated production and testing platform capable of meeting both clinical and commercial needs and we are committing substantial resources in this area. Important features of our capabilities are set forth below.
| • | Our propriety platform for AAV production generates high quality rAAV vectors with high packaging fidelity, high infectivity and low empty particles across multiple serotypes. |
| • | Our AAV production system generates high volumetric productivities and has achieved more than 10-fold improvement in productivity compared to other manufacturing formats. |
| • | We have adapted our herpes simplex virus, or HSV, helper manufacturing system to multiple vendors’ single use bioreactors, demonstrating robustness and flexibility while removing scale and format limitations attendant with adherent cell culture. |
| • | We have optimized purification and formulation activities to yield multiple rAAV serotypes in a dose-ready form with exceptional purity at previously unattainable genomic concentrations. |
| • | Our integrated testing platform has generated over 35 product-specific characterization assays that have been successfully transferred for the evaluation of HSV helpers and AAV vectors at contract testing organizations. |
| • | The robust cell substrates we employ are well characterized and have been reviewed in several regulatory submissions in the U.S., Canada, Israel and Europe. |
| • | Our ability to successfully transfer the technology to multiple contract manufacturing organizations as well as collaboration partners demonstrates the robustness of our manufacturing process. |
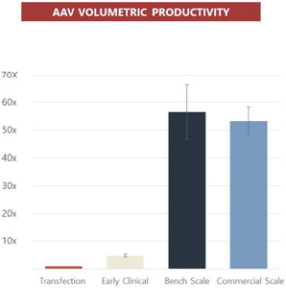
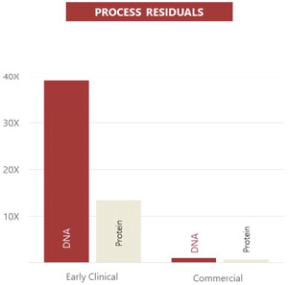
Taken together, we believe that the efficiency, productivity, scalability, characterization and regulatory definition of our proprietary rAAV manufacturing platform uniquely position us to quickly transition from early phase human clinical trials to late phase, BLA-enabling data in all our clinical programs. We are currently at commercial scale for our orphan ophthalmology programs due to our high productivity that enable us to achieve thousands of doses from a small bioreactor. As such, in the near term, it is more efficient for us to pursue a hybrid strategy where we have developed and optimized the manufacturing process and leverage a CDMO’s investment in capital equipment when needed. Productivity (below, left) has increased 55-fold between early clinical and commercial processes at scale, while process residuals (below, right) have decreased nearly 40-fold (DNA) or greater than 10-fold (protein).
|
|
7