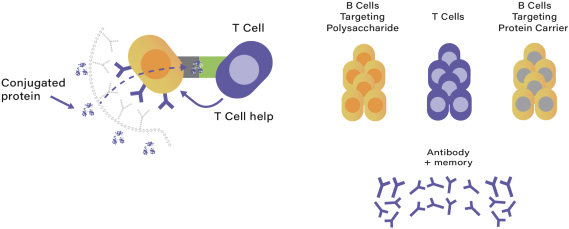
President and Chief Financial Officer at Neoforma, Inc., a provider of supply-chain management solutions for the healthcare industry. Mr. Guggenhime currently serves on the board of
directors of Metacrine, Inc., a privately held biotechnology company. Mr. Guggenhime holds a B.A. in International Politics and Economics from Middlebury College and an M.B.A. from the J.L. Kellogg Graduate School of Management at Northwestern
University.
Jim Wassil, M.S., M.B.A. Mr. Wassil has served as our Chief Operating Officer since December 1, 2019. From May
2015 to December 2019, Mr. Wassil served as Vice President and Global Health and Value Business Unit Lead, Vaccines at Pfizer Inc., a multinational pharmaceutical company. From August 2008 to May 2015, Mr. Wassil served as Head, Global Product
Development Meningococcal Vaccines and Head, U.S. Marketing for Meningococcal Vaccines at Novartis AG, a multinational pharmaceutical company. Prior to joining Novartis, Mr. Wassil served as Senior Director, International Marketing at Merck
& Co., Inc., a multinational pharmaceutical company. Mr. Wassil is a member of the Infectious Diseases Society of America. Mr. Wassil holds a B.S. in Chemistry/Biology from the University of Notre Dame and a M.S. in BioOrganic Chemistry and an
M.B.A. from Lehigh University.
Paul Sauer, M.B.A. Mr. Sauer has served as our Senior Vice President, Process
Development and Manufacturing since April 2016. From January 2015 to March 2016, Mr. Sauer served as a Principal at Sauer Biotech Consulting, a development and manufacturing consulting services firm. From July 2011 to December 2014,
Mr. Sauer served as Vice President, Process Sciences and Manufacturing at Igencia Biotherapeutics, Inc., a biotechnology company. Mr. Sauer holds a B.S. in Genetics and a B.A. in Psychology from the University of California, Davis and an
M.B.A. from Santa Clara University.
Jane Wright-Mitchell, PharmD, J.D. Ms. Wright-Mitchell has served as our
General Counsel since January 2019. From November 2017 to December 2018, Ms. Wright-Mitchell served as Chief Legal Officer at Steep Hill, Inc., a cannabis testing and analytics company. From July 2014 to November 2017, Ms. Wright-Mitchell
served as Chief Legal Officer at AcelRx Pharmaceuticals, Inc., a pharmaceutical company. Ms. Wright-Mitchell holds a B.S. in Biological Sciences from Clemson University, a PharmD from the University of Illinois at Chicago and a J.D. from
Chicago-Kent College of Law, Illinois Institute of Technology.
Jeff Fairman, Ph.D. Mr. Fairman is our co-founder and has served as our Vice President, Research since December 2013. From July 2011 to December 2013, Mr. Fairman served as Vice President, Research at Colby Pharmaceuticals, a biopharmaceutical
company. Mr. Fairman also founded Juvaris BioTherapeutics, Inc., a biopharmaceutical company, and served as its Vice President, Research from February 2002 to September 2011. Mr. Fairman is a member of the American Association of
Immunologists and the Infectious Diseases Society of America. Mr. Fairman holds a B.S. in Chemistry from Northwest Missouri State University and a Ph.D. in Chemistry from the University of Arkansas.
Non-Employee Directors
Moncef Slaoui, Ph.D. Dr. Slaoui has served on our board of directors since July 2017 and has served as Chairman of
the Board since May 2018. Dr. Slaoui currently serves as a Partner at Medicxi, a venture capital firm. In May 2020, Dr. Slaoui was appointed as chief advisor to the White House’s Operation Warp Speed initiative, the administration’s
national program to accelerate the development, manufacturing, and distribution of COVID-19 vaccines, therapeutics, and diagnostics. From June 2009 to June 2017, Dr. Slaoui served as the Chairman of Vaccines at GlaxoSmithKline plc, a
multinational pharmaceutical company, and from June 2003 to June 2006, he served as head of Worldwide Business Development at GlaxoSmithKline. Dr. Slaoui currently serves on the boards of directors of private biotechnology companies. From July
2017 to May 2020, Dr. Slaoui served on the board of directors of Moderna, Inc., a biotechnology company, and from April 2020 to May 2020, Dr. Slaoui served on the board of directors of Lonza Group Ltd. From 1984 to 1988, Dr. Slaoui served as a
professor of Immunology at the University of Mons, Belgium. Dr. Slaoui holds a Ph.D. in Molecular Biology and Immunology from the Université Libre de Bruxelles, Belgium. Dr. Slaoui was selected to serve on our board of directors
because of his depth of vaccine industry and public company experience.
146