UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-Q
|
|
|
|
|
☒
|
QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
|
|
For the quarterly period ended March 31, 2020
OR
|
|
|
|
|
◻
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
|
|
For the transition period from _________________ to _______________________
Commission file number: 001‑37544
ARMATA PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
|
|
|
|
Washington
|
91‑1549568
|
|
(State or other jurisdiction of
|
(I.R.S. Employer Identification Number)
|
|
incorporation or organization)
|
|
|
|
|
|
4503 Glencoe Avenue
|
|
|
Marina del Rey, CA
|
90292
|
|
(Address of principal executive offices)
|
(Zip Code)
|
Registrant’s telephone number, including area code: (310) 665-2928
Securities registered pursuant to Section 12(b) of the Act:
|
|
|
|
|
Title of each class
|
Trading Symbol(s)
|
Name of each exchange on which registered
|
|
Common Stock, $0.01 par value per share
|
ARMP
|
NYSE American
|
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes ☒ No ◻
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Yes ☒ No ◻
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company as defined in Rule 12b‑2 of the Exchange Act. See the definitions of “large accelerated filer,” “accelerated filer”, “smaller reporting company” and “emerging growth company” in Rule 12b‑2 of the Exchange Act.
|
|
|
|
Large accelerated filer ◻
|
Accelerated filer ◻
|
|
Non-accelerated filer ☒
|
Smaller reporting company ☒
|
|
|
Emerging growth company ◻
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ◻
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b‑2 of the Exchange Act).
Yes ◻ No ☒
The number of shares of the registrant’s Common Stock, par value $0.01 per share, outstanding at May 6, 2020 was 18,644,693.
Armata Pharmaceuticals, Inc.
Consolidated Balance Sheets
|
|
|
|
|
|
|
|
|
|
|
|
March 31, 2020
|
|
December 31, 2019
|
|
|
|
|
(unaudited)
|
|
|
|
|
|
Assets
|
|
|
|
|
|
|
|
|
Current assets
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
24,209,000
|
|
$
|
6,033,000
|
|
|
Award receivable
|
|
|
1,000,000
|
|
|
—
|
|
|
Prepaid expenses and other current assets
|
|
|
567,000
|
|
|
622,000
|
|
|
Total current assets
|
|
|
25,776,000
|
|
|
6,655,000
|
|
|
Restricted cash
|
|
|
600,000
|
|
|
700,000
|
|
|
Property and equipment, net
|
|
|
2,005,000
|
|
|
2,187,000
|
|
|
Operating lease right-of-use asset
|
|
|
1,839,000
|
|
|
2,028,000
|
|
|
In-process research and development
|
|
|
10,256,000
|
|
|
10,256,000
|
|
|
Goodwill
|
|
|
3,490,000
|
|
|
3,490,000
|
|
|
Other assets
|
|
|
136,000
|
|
|
135,000
|
|
|
Total assets
|
|
$
|
44,102,000
|
|
$
|
25,451,000
|
|
|
|
|
|
|
|
|
|
|
|
Liabilities and stockholders’ equity
|
|
|
|
|
|
|
|
|
Current liabilities
|
|
|
|
|
|
|
|
|
Accounts payable and accrued liabilities
|
|
$
|
1,447,000
|
|
$
|
1,278,000
|
|
|
Accrued compensation
|
|
|
1,209,000
|
|
|
1,323,000
|
|
|
Deferred award liability
|
|
|
859,000
|
|
|
—
|
|
|
Deferred asset acquisition consideration
|
|
|
1,476,000
|
|
|
970,000
|
|
|
Current portion of operating lease liabilities
|
|
|
1,366,000
|
|
|
1,308,000
|
|
|
Total current liabilities
|
|
|
6,357,000
|
|
|
4,879,000
|
|
|
Operating lease liabilities, net of current portion
|
|
|
1,186,000
|
|
|
1,555,000
|
|
|
Deferred asset acquisition consideration, net of current portion
|
|
|
—
|
|
|
1,347,000
|
|
|
Deferred tax liability
|
|
|
3,077,000
|
|
|
3,077,000
|
|
|
Total liabilities
|
|
|
10,620,000
|
|
|
10,858,000
|
|
|
|
|
|
|
|
|
|
|
|
Stockholders’ equity
|
|
|
|
|
|
|
|
|
Common stock, $0.01 par value; 217,000,000 shares authorized; 18,644,693 and 9,922,758 shares issued and outstanding at March 31, 2020 and December 31, 2019, respectively.
|
|
|
186,000
|
|
|
99,000
|
|
|
Additional paid-in capital
|
|
|
195,895,000
|
|
|
172,015,000
|
|
|
Accumulated deficit
|
|
|
(162,599,000)
|
|
|
(157,521,000)
|
|
|
Total stockholders’ equity
|
|
|
33,482,000
|
|
|
14,593,000
|
|
|
Total liabilities and stockholders’ equity
|
|
$
|
44,102,000
|
|
$
|
25,451,000
|
|
See accompanying condensed notes to consolidated financial statements.
Armata Pharmaceuticals, Inc.
Consolidated Statements of Operations
|
|
|
|
|
|
|
|
|
|
|
|
Three Months Ended
|
|
|
|
|
March 31,
|
|
|
|
|
2020
|
|
2019
|
|
|
|
|
|
(unaudited)
|
|
|
(unaudited)
|
|
|
Operating expenses
|
|
|
|
|
|
|
|
|
Research and development
|
|
|
2,750,000
|
|
|
2,061,000
|
|
|
General and administrative
|
|
|
2,171,000
|
|
|
1,380,000
|
|
|
Loss from operations
|
|
|
4,921,000
|
|
|
3,441,000
|
|
|
Other income (expense)
|
|
|
|
|
|
|
|
|
Interest income
|
|
|
2,000
|
|
|
48,000
|
|
|
Interest expense
|
|
|
(159,000)
|
|
|
(306,000)
|
|
|
Change in fair value of derivative liabilities
|
|
|
—
|
|
|
(40,000)
|
|
|
Total other income (expense), net
|
|
|
(157,000)
|
|
|
(298,000)
|
|
|
Net loss
|
|
$
|
(5,078,000)
|
|
$
|
(3,739,000)
|
|
|
Per share information:
|
|
|
|
|
|
|
|
|
Net loss per share, basic and diluted
|
|
$
|
(0.49)
|
|
$
|
(0.80)
|
|
|
Weighted average shares outstanding, basic and diluted
|
|
|
10,451,746
|
|
|
4,652,777
|
|
See accompanying condensed notes to consolidated financial statements.
Armata Pharmaceuticals, Inc.
Consolidated Statements of Stockholders’ Equity
Three Months Ended March 31, 2020 and 2019
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stockholders’ Equity
|
|
|
|
Common Stock
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
|
Total
|
|
|
|
|
|
|
|
|
Paid-in
|
|
Accumulated
|
|
Stockholders’
|
|
|
|
Shares
|
|
Amount
|
|
Capital
|
|
Deficit
|
|
Equity
|
|
Balances, December 31, 2018
|
|
5,069,633
|
|
$
|
51,000
|
|
$
|
145,685,000
|
|
$
|
(138,042,000)
|
|
$
|
7,694,000
|
|
Net loss
|
|
—
|
|
|
—
|
|
|
—
|
|
|
(3,739,000)
|
|
|
(3,739,000)
|
|
Balances, March 31, 2019
|
|
5,069,633
|
|
$
|
51,000
|
|
$
|
145,685,000
|
|
$
|
(141,781,000)
|
|
$
|
3,955,000
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balances, December 31, 2019
|
|
9,922,758
|
|
$
|
99,000
|
|
$
|
172,015,000
|
|
$
|
(157,521,000)
|
|
$
|
14,593,000
|
|
Sale of common stock, net of issuance costs
|
|
8,710,800
|
|
|
87,000
|
|
|
22,755,000
|
|
|
—
|
|
|
22,842,000
|
|
Exercises of warrants
|
|
14,464
|
|
|
—
|
|
|
81,000
|
|
|
—
|
|
|
81,000
|
|
Forfeiture of restricted stock awards
|
|
(3,329)
|
|
|
—
|
|
|
—
|
|
|
—
|
|
|
—
|
|
Stock-based compensation
|
|
—
|
|
|
—
|
|
|
1,044,000
|
|
|
—
|
|
|
1,044,000
|
|
Net loss
|
|
—
|
|
|
—
|
|
|
—
|
|
|
(5,078,000)
|
|
|
(5,078,000)
|
|
Balances, March 31, 2020
|
|
18,644,693
|
|
$
|
186,000
|
|
$
|
195,895,000
|
|
$
|
(162,599,000)
|
|
$
|
33,482,000
|
See accompanying condensed notes to consolidated financial statements.
Armata Pharmaceuticals, Inc.
Consolidated Statements of Cash Flows
|
|
|
|
|
|
|
|
|
|
|
Three Months Ended March 31,
|
|
|
|
2020
|
|
2019
|
|
|
|
(unaudited)
|
|
(unaudited)
|
|
Operating activities:
|
|
|
|
|
|
|
|
Net loss
|
|
$
|
(5,078,000)
|
|
$
|
(3,739,000)
|
|
Adjustments required to reconcile net loss to net cash used in operating activities:
|
|
|
|
|
|
|
|
Depreciation
|
|
|
295,000
|
|
|
348,000
|
|
Stock-based compensation
|
|
|
1,044,000
|
|
|
—
|
|
Non-cash interest expense
|
|
|
159,000
|
|
|
306,000
|
|
Change in fair value of derivative liability
|
|
|
—
|
|
|
40,000
|
|
Changes in operating assets and liabilities:
|
|
|
|
|
|
|
|
Award receivable
|
|
|
(1,000,000)
|
|
|
—
|
|
Accounts payable and accrued liabilities
|
|
|
(195,000)
|
|
|
429,000
|
|
Accrued compensation
|
|
|
(114,000)
|
|
|
—
|
|
Deferred rent and lease liabilities, net
|
|
|
(122,000)
|
|
|
(84,000)
|
|
Deferred award liability
|
|
|
859,000
|
|
|
—
|
|
Prepaid expenses and other current assets
|
|
|
(80,000)
|
|
|
241,000
|
|
Net cash used in operating activities
|
|
|
(4,232,000)
|
|
|
(2,459,000)
|
|
Investing activities:
|
|
|
|
|
|
|
|
Purchases of property and equipment
|
|
|
(104,000)
|
|
|
(142,000)
|
|
Net cash used in investing activities
|
|
|
(104,000)
|
|
|
(142,000)
|
|
Financing activities:
|
|
|
|
|
|
|
|
Payment of deferred consideration for asset acquisition
|
|
|
(1,000,000)
|
|
|
(1,000,000)
|
|
Proceeds from sale of common stock, net of offering costs
|
|
|
23,331,000
|
|
|
—
|
|
Proceeds from exercise of warrants
|
|
|
81,000
|
|
|
—
|
|
Net cash provided by (used in) financing activities
|
|
|
22,412,000
|
|
|
(1,000,000)
|
|
Net increase (decrease) in cash, cash equivalents and restricted cash
|
|
|
18,076,000
|
|
|
(3,601,000)
|
|
Cash, cash equivalents and restricted cash, beginning of period
|
|
|
6,733,000
|
|
|
10,463,000
|
|
Cash, cash equivalents and restricted cash, end of period
|
|
$
|
24,809,000
|
|
$
|
6,862,000
|
|
Supplemental schedule of non-cash investing and financing activities:
|
|
|
|
|
|
|
|
Property and equipment included in accounts payable
|
|
$
|
9,000
|
|
$
|
—
|
|
Unpaid offering costs
|
|
$
|
471,000
|
|
$
|
—
|
The following table provides a reconciliation of cash, cash equivalents, and restricted cash reported within the consolidated balance sheets that sum to the total of the same amounts shown in the consolidated statement of cash flows:
|
|
|
|
|
|
|
|
|
|
|
Three Months Ended March 31,
|
|
|
|
2020
|
|
2019
|
|
Cash and cash equivalents
|
|
$
|
24,209,000
|
|
$
|
6,162,000
|
|
Restricted cash
|
|
|
600,000
|
|
|
700,000
|
|
Cash, cash equivalents and restricted cash
|
|
$
|
24,809,000
|
|
$
|
6,862,000
|
See accompanying condensed notes to consolidated financial statements.
Armata Pharmaceuticals, Inc.
Condensed Notes to Consolidated Financial Statements
(Unaudited)
1. Organization and Description of the Business
Armata Pharmaceuticals, Inc. (“Armata”, and together with its subsidiaries referred to herein as, the “Company”) is a clinical-stage biotechnology company focused on the development of precisely targeted bacteriophage therapeutics for the treatment of antibiotic-resistant infections using its proprietary bacteriophage-based technology. The Company was created as a result of a business combination between C3J Therapeutics, Inc. (“C3J”), a Washington company, and AmpliPhi Biosciences Corporation (“AmpliPhi”) that closed on May 9, 2019, where Ceres Merger Sub, Inc., a wholly owned subsidiary of AmpliPhi, merged with and into C3J (the ”Merger”), with C3J surviving the Merger as a wholly owned subsidiary of AmpliPhi. In the Merger, each share of C3J common stock outstanding immediately prior to the Merger was converted into the right to receive approximately .6906 shares of AmpliPhi common stock. The shares were then adjusted further to account for a reverse split of AmpliPhi common stock at a reverse split ratio of 1‑for‑14. All share and per share amounts have been retrospectively adjusted to give effect to the exchange of C3J common stock and the reverse split of AmpliPhi common stock.
Immediately prior to the closing of the Merger, AmpliPhi changed its name to Armata Pharmaceuticals, Inc. Armata’s common stock is traded on the NYSE American exchange under the ticker symbol “ARMP.”
Immediately following the Merger, certain existing C3J shareholders purchased $10.0 million in Armata common stock. After the Merger and such concurrent private placement, the former C3J security holders owned approximately 76% of the aggregate number of shares of Armata’s common stock and the security holders of AmpliPhi as of immediately prior to the Merger owned approximately 24% of the aggregate number of shares of Armata’s common stock. In addition, upon closing of the Merger, five of the seven members of the board of directors were appointed by C3J.
In connection with the Merger, C3J was considered the accounting acquirer of AmpliPhi because C3J’s shareholders retained a majority control of ownership of the Company subsequent to the Merger. In addition, the seven-member board of directors of the combined company include five members established by C3J. Therefore, the historical financial statements presented herein prior to the closing of the Merger are the historical financial statements of C3J.
C3J’s predecessor, C3 Jian, Inc., was incorporated under the laws of the State of California on November 4, 2005. On February 26, 2016, as part of a reorganization transaction, C3 Jian, Inc. merged with a wholly owned subsidiary of C3J, and as part of this process, C3 Jian, Inc. was converted to a limited liability company organized under the laws of the State of California named C3 Jian, LLC. Prior to the Merger, C3J was privately held and was financed principally through a series of equity financings.
2. Liquidity
The Company has prepared its consolidated financial statements on a going concern basis, which assumes that the Company will realize its assets and satisfy its liabilities in the normal course of business. However, the Company has incurred net losses since its inception and has negative operating cash flows. These circumstances raise substantial doubt about the Company’s ability to continue as a going concern. The accompanying financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classifications of liabilities that may result from the outcome of the uncertainty concerning the Company’s ability to continue as a going concern.
As described in more detail in Note 8, on March 27, 2020, the Company completed a private placement transaction and sold to Innoviva Inc. (“Innoviva”) 8,710,800 newly issued shares of the Company’s common stock and warrants to purchase 8,710,800 shares of common stock, with an exercise price per share of $2.87 (the “Private Placement”). Each share of common stock was sold together with one common warrant granting the warrant holder the right to purchase an
additional share of common stock at $2.87 per share. The Private Placement was closed in two tranches raising total gross proceeds of $25.0 million.
As of March 31, 2020, the Company had cash and cash equivalents of $24.2 million. Considering the Company’s current cash resources, management believes the Company’s existing resources will be sufficient to fund the Company’s planned operations into the second quarter of 2021. For the foreseeable future, the Company’s ability to continue its operations is dependent upon its ability to obtain additional capital.
Management plans to raise additional capital through equity offerings, debt financings, or other capital sources, including potential collaborations, grants, licensing of intellectual property, and other similar arrangements. While management believes this plan to raise additional funds will alleviate the conditions that raise substantial doubt, these plans are not entirely within its control and cannot be assessed as being probable of occurring. The Company’s ability to raise additional capital may be adversely impacted by potential worsening global economic conditions and the recent disruptions to, and volatility in, financial markets in the United States and worldwide resulting from the ongoing COVID-19 pandemic. The Company may not be able to secure additional financing in a timely manner or on favorable terms, if at all. Furthermore, if the Company issues equity securities to raise additional funds, its existing stockholders may experience dilution, and the new equity securities may have rights, preferences and privileges senior to those of the Company’s existing stockholders. If the Company raises additional funds through collaboration, licensing or other similar arrangements, it may be necessary to relinquish valuable rights to its potential products on terms that are not favorable to the Company. If the Company is unable to raise capital when needed or on attractive terms, it would be forced to delay, reduce or eliminate its research and development programs or other operations. If any of these events occur, the Company’s ability to achieve the development and commercialization goals would be adversely affected.
3. Significant Accounting Policies
Basis of Presentation
The consolidated financial statements include the accounts of Armata and its wholly owned subsidiaries. All significant intercompany accounts and transactions have been eliminated. The accompanying unaudited consolidated financial statements of the Company should be read in conjunction with the audited financial statements and accompanying notes thereto as of and for the year ended December 31, 2019 included in the Company’s Form 10-K, filed with the U.S. Securities and Exchange Commission on March 19, 2020. The accompanying unaudited financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America (“U.S. GAAP”) for interim financial statements. Any reference in the Notes to applicable guidance is meant to refer to authoritative U.S. GAAP as found in the Accounting Standards Codification (“ASC”) and Accounting Standards Update (“ASU”) of the Financial Accounting Standards Board (“FASB”).
In the opinion of management, the accompanying consolidated financial statements include all adjustments that are of a normal and recurring nature and that are necessary for the fair presentation of the Company’s financial position and the results of its operations and cash flows for the periods presented. Interim results are not necessarily indicative of results for the full year or any future period.
Use of Estimates
The preparation of consolidated financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the amounts reported in its consolidated financial statements and accompanying notes. On an ongoing basis, management evaluates these estimates and judgments, which are based on historical and anticipated results and trends, and on various other assumptions that management believes to be reasonable under the circumstances. By their nature, estimates are subject to an inherent degree of uncertainty and, as such, actual results may differ from management’s estimates.
Fair Value of Financial Instruments
The carrying amounts of cash equivalents, other current assets, accounts payable, and accrued liabilities approximate fair value because of the short-term nature of these instruments.
In-Process Research and Development (“IPR&D”)
IPR&D assets are intangible assets with indefinite lives and are not subject to amortization. The Company’s IPR&D assets represent capitalized incomplete research projects that the Company acquired through the Merger. Such assets are initially measured at their acquisition-date fair values and are subject to impairment testing at least annually until completion or abandonment of research and development efforts associated with the projects. Upon successful completion of each project, the Company makes a determination as to the then remaining useful life of the intangible asset and begins amortization.
Goodwill
Goodwill, which has an indefinite useful life, represents the excess of purchase consideration over fair value of net assets acquired. The Company’s goodwill as of March 31, 2020 is associated with AmpliPhi’s business prior to the Merger. Goodwill is not subject to amortization and is required to be tested for impairment at least on an annual basis. The Company tests goodwill for impairment as of December 31 of each year. The Company determines whether goodwill may be impaired by comparing the carrying value of the single reporting unit, including goodwill, to the fair value of the reporting unit. If the fair value is less than the carrying amount, a more detailed analysis is performed to determine whether goodwill is impaired. The impairment loss, if any, is measured as the excess of the carrying value of the goodwill over the implied fair value of the goodwill and is recorded in the Company’s consolidated statements of operations.
Derivative Liabilities
Derivative liabilities are accounted for in accordance with the applicable accounting guidance provided in ASC 815 – Derivatives and Hedging based on the specific terms of the agreements. Derivative liabilities are recorded at fair value at each reporting period with any change in fair value recognized as a component of change in fair value of asset acquisition derivative liability in the consolidated statements of operations. The Company has a zero derivative liability balance at March 31, 2020 as the liability of $1.1 million at December 31, 2018 was settled upon the Merger in May 2019.
Basic and Diluted Net Loss per Share
Net earnings or loss per share (“EPS”) is calculated in accordance with the applicable accounting guidance provided in ASC 260, Earnings per Share. The Company uses the two-class method for the computation and presentation of net income (loss) per common share attributable to common stockholders. The two-class method is an earnings allocation formula that calculates basic and diluted net income (loss) per common share for each class of common stock separately based on dividends declared and participation rights in undistributed earnings as if all such earnings had been distributed during the period. Under the two-class method, warrants issued to Innoviva in connection with the Private Placement (Note 2) is assumed to participate in undistributed earnings on an as-exercised basis, in accordance with the warrant agreement. Undistributed net losses are allocated entirely to common shareholders since the participating security has no contractual obligation to share in the losses.
Accordingly, basic income or loss per share is calculated by dividing net income or loss by the weighted-average number of common shares outstanding, or using the two-class method, whichever is more dilutive. Diluted net income loss per share is computed using the more dilutive of the treasury stock method which reflects the potential dilution that would occur if securities or other contracts to issue common stock were exercised or converted to common stock, or the two-class method.
The calculation of diluted loss per share requires that, to the extent the average market price of the underlying shares for the reporting period exceeds the exercise price of the warrants, and the presumed exercise of such securities are dilutive to net loss per share for the period, an adjustment to net loss available to common stockholders used in the calculation is required to remove the change in fair value of the warrants from the numerator for the period. Likewise, an adjustment to the denominator is required to reflect the related dilutive shares, if any, under the treasury stock method.
Grants and Awards
In applying the provisions of ASC Topic 606, Revenue from Contracts with Customers (“ASC 606”), Armata has determined that grants and awards are out of the scope of ASC 606 because the funding entities do not meet the definition of a “customer”, as defined by ASC 606, as there is not considered to be a transfer of control of goods or services. With respect to each grant or award, the Company determines if it has a collaboration in accordance with ASC Topic 808, Collaborative Arrangements (“ASC 808”). To the extent the grant or award is within the scope of ASC 808, the Company recognizes amounts received as a contra-expense, as opposed to revenue, on the consolidated statement of operations when the related research and development expenses are incurred. Armata also considers the guidance in ASC Topic 730, Research and Development (“ASC 730”), which requires an assessment, at the inception of the grant or award, of whether the agreement is a liability. If Armata is obligated to repay funds received regardless of the outcome of the related research and development activities, then Armata is required to estimate and recognize that liability. Alternatively, if Armata is not required to repay the funds, then payments received are recorded as a contra-expense as the expenses are incurred.
Deferred award liability represents award funds received or receivable for which the allowable expenses have not yet been incurred as of the balance sheet date.
Research and Development Expenses
Research and development (“R&D”) costs consist primarily of direct and allocated salaries, incentive compensation, stock-based compensation and other personnel-related costs, facility costs, and third-party services. Third-party services include studies and clinical trials conducted by clinical research organizations. R&D activities are expensed as incurred. The Company records accruals for estimated ongoing clinical trial expenses. When evaluating the adequacy of the accrued liabilities, the Company analyzes progress of the studies, including the phase or completion of events, invoices received and contracted costs. Judgments and estimates are made in determining the accrued balances at the end of the reporting period.
Recent Accounting Pronouncements Not Yet Adopted
In June 2016, the FASB issued ASU 2016-13, Financial Instruments - Credit Losses (Topic 326), Measurement of Credit Losses on Financial Instruments. The standard amends the impairment model by requiring entities to use a forward-looking approach based on expected losses to estimate credit losses for most financial assets and certain other instruments that aren’t measured at fair value through net income. For available-for-sale debt securities, entities will be required to recognize an allowance for credit losses rather than a reduction in carrying value of the asset. Entities will no longer be permitted to consider the length of time that fair value has been less than amortized cost when evaluating when credit losses should be recognized. This new guidance is effective for calendar-year smaller reporting public entities in the first quarter of 2023. The Company is currently evaluating the impact of this ASU and does not expect that adoption of this standard will have a material impact on its consolidated financial statements or related disclosures.
In December 2019, the FASB issued ASU 2019-12, Income Taxes (“ASC 740”), which simplifies the accounting for income taxes by eliminating certain exceptions to the guidance in ASC 740 related to the approach for intra-period tax allocation, the methodology for calculating income taxes in an interim period and the recognition of deferred tax liabilities for outside basis differences. The new guidance also simplifies aspects of the accounting for franchise taxes and enacted changes in tax laws or rates and clarifies the accounting for transactions that result in a step-up in the tax basis of goodwill. The guidance is effective for calendar-year public business entities in 2021 and interim periods within that year.
Early adoption is permitted. The Company does not expect adoption of this new guidance will have a material impact on its consolidated financial statements or related disclosures.
Recently Adopted Accounting Standards
In November 2018, FASB issued ASU 2018-18, Clarifying the Interaction between Topic 808 and Topic 606. The objective of the standard is to clarify the interaction between Topic 808, Collaborative Arrangements, and Topic 606, Revenue from Contracts with Customers. Currently, Topic 808 does not provide comprehensive recognition or measurement guidance for collaborative arrangements, and the accounting for those arrangements is often based on an analogy to other accounting literature or an accounting policy election. Similarly, aspects of Topic 606 have resulted in uncertainty in practice about the effect of the revenue standard and credit loss standard on the accounting for collaborative arrangements. The standard became effective for the Company for fiscal periods beginning on January 1, 2020. The adoption of this ASU did not have an impact on the Company’s financial condition, results of operations, cash flows, or financial statement disclosures.
4. Fair Value Measurements
The guidance regarding fair value measurements prioritizes the inputs used in measuring fair value and establishes a three-tier value hierarchy that distinguishes among the following:
|
·
| |
Level 1—Valuations based on unadjusted quoted prices in active markets for identical assets or liabilities that the Company has the ability to access. |
|
·
| |
Level 2—Valuations based on quoted prices for similar assets or liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets that are not active and models for which all significant inputs are observable, either directly or indirectly. |
|
·
| |
Level 3—Valuations based on inputs that are unobservable and significant to the overall fair value measurement. |
The Company estimates the fair values of derivative liabilities utilizing Level 3 inputs. No derivative liabilities have been transferred between the classification levels. Estimating the fair values of derivative liabilities requires the use of significant and subjective inputs that may, and are likely to, change over the duration of the instrument with related changes in internal and external market factors.
The following table sets forth a summary of changes in the fair value of the Company’s liabilities during the three months ended March 31, 2019:
|
|
|
|
|
|
|
|
|
Asset
|
|
|
|
|
Acquisition
|
|
|
|
|
Derivative
|
|
|
|
|
Liability
|
|
|
Balance, December 31, 2018
|
|
$
|
1,117,000
|
|
|
Changes in estimated fair value
|
|
|
40,000
|
|
|
Balance, March 31, 2019
|
|
$
|
1,157,000
|
|
We estimated the fair value of this derivative by forecasting the timing and likelihood of the events occurring and discounting the probability adjusted payments using an appropriate discount based on market interest rates and our own non-performance risk as required by ASC 820 – Fair Value Measurement. There is no longer a potential payment requirement associated with the derivative liability subsequent to the Merger. Accordingly, the fair value of the derivative liability was reduced to zero in the second quarter of 2019 with the associated change recorded in other income.
5. The Merger
On May 9, 2019, the Company completed the Merger (see Note 1). On the date of the Merger, AmpliPhi had, and the Company currently has, IPR&D related to the development of AP-SA01, a phage combination for the treatment of Staphylococcus aureus infections, and had tested such product in patients through single-patient expanded access guidelines established by U.S. and Australian regulatory agencies. Further, AmpliPhi had, and the Company currently has, a workforce that is considered to have the necessary skills, knowledge, and experience to perform a process, that when applied to IPR&D is critical to the ability to convert it into outputs. Based on this evaluation, the Company determined that the Merger should be accounted for as a business combination pursuant to Financial Accounting Standards Board Accounting Standards Codification Topic 805, Business Combinations (“ASC 805”).
In connection with the Merger, the Company allocated the total purchase consideration of $10.7 million in stock to the net assets and liabilities acquired, including goodwill of $3.5 million, identifiable intangible assets of $10.3 million and related deferred tax liability of $3.1 million, based on their respective fair values at the acquisition date. The Company recognizes deferred tax liabilities for indefinite-lived intangible assets in accordance with ASC 740, Income Taxes.
In addition, the Company incurred and expensed costs directly related to the Merger totaling approximately $1.1 million, of which approximately zero and $0.6 million was incurred in the three months ended March 31, 2020 and March 31, 2019, respectively, and is included in general and administrative expenses in the consolidated statement of operations.
Since the closing date of the Merger, the results of AmpliPhi’s operations have been included in the Company’s consolidated financial statements. Selected amounts related to AmpliPhi’s business included in the Company’s consolidated statements of operations for the three months ended March 31, 2020, are as follows:
|
|
|
|
|
Three Months Ended March 31,
|
|
|
2020
|
|
Research and development expenses
|
$ 122,000
|
|
General and administrative expenses
|
260,000
|
|
Net loss
|
$ 382,000
|
6. Net Loss per Share
The following outstanding securities at March 31, 2020 and 2019 have been excluded from the computation of diluted weighted average shares outstanding for the three months ended March 31, 2020 and 2019, as they would have been anti-dilutive:
|
|
|
|
|
|
|
|
|
Three Months Ended
|
|
|
|
March 31,
|
|
|
|
2020
|
|
2019
|
|
Options
|
|
1,365,764
|
|
136,463
|
|
Restricted stock awards
|
|
340,164
|
|
416,856
|
|
Warrants
|
|
10,547,363
|
|
—
|
|
Total
|
|
12,253,291
|
|
553,319
|
7. Balance Sheet Details
Property and Equipment
Property and equipment as of March 31, 2020 and December 31, 2019 consisted of the following:
|
|
|
|
|
|
|
|
|
|
|
March 31, 2020
|
|
December 31, 2019
|
|
Laboratory equipment
|
|
$
|
6,127,000
|
|
$
|
6,047,000
|
|
Furniture and fixtures
|
|
|
646,000
|
|
|
646,000
|
|
Office and computer equipment
|
|
|
334,000
|
|
|
323,000
|
|
Leasehold improvements
|
|
|
3,352,000
|
|
|
3,329,000
|
|
Total
|
|
|
10,459,000
|
|
|
10,345,000
|
|
Less: accumulated depreciation
|
|
|
(8,454,000)
|
|
|
(8,158,000)
|
|
Property and equipment, net
|
|
$
|
2,005,000
|
|
$
|
2,187,000
|
Depreciation expense totaled $295,000 and $348,000 for the three months ended March 31, 2020 and 2019, respectively.
8. Stockholders’ Equity
Private Investment
On January 27, 2020, the Company entered into the Securities Purchase Agreement with Innoviva, pursuant to which the Company agreed to issue and sell to Innoviva, in a Private Placement, 8,710,800 newly issued shares of the Company’s common stock and warrants to purchase 8,710,800 shares of common stock, with an exercise price per share of $2.87. Each share of common stock was sold together with one common warrant granting the warrant holder the right to purchase an additional share of common stock at $2.87 per share. The Private Placement occurred in two tranches. The first closing occurred on February 12, 2020, at which time Innoviva purchased 993,139 Common Units in exchange for an aggregate gross cash payment of approximately $2.8 million. On March 27, 2020, the second closing occurred subsequent to shareholder approval, at which time Innoviva purchased 7,717,661 Common Units in exchange for aggregate gross proceeds of $22.2 million.
The warrants expire five years from the issuance date. The Company reviewed the authoritative accounting guidance and determined that the warrants meet the criteria to be accounted for as permanent equity.
Warrants
At March 31, 2020, outstanding warrants to purchase shares of common stock are as follows:
|
|
|
|
|
|
|
|
Shares Underlying
|
|
|
|
|
|
|
Outstanding
|
|
Exercise
|
|
Expiration
|
|
Warrants
|
|
Price
|
|
Date
|
|
1,991
|
|
$
|
567.00
|
|
March 31, 2021
|
|
597,881
|
|
$
|
21.00
|
|
May 10, 2022
|
|
1,235,491
|
|
$
|
5.60
|
|
October 16, 2023
|
|
993,139
|
|
$
|
2.87
|
|
February 12, 2025
|
|
7,717,661
|
|
$
|
2.87
|
|
March 27, 2025
|
|
1,200
|
|
$
|
1,680.00
|
|
None
|
|
10,547,363
|
|
|
|
|
|
9. Equity Incentive Plans
Stock Award Plans
The Company maintains a 2016 Equity Incentive Plan (the “2016 Plan”), which provides for the issuance of incentive share awards in the form of non-qualified and incentive stock options, stock appreciation rights, restricted stock awards, restricted stock unit awards and performance-based stock awards. The awards may be granted by the Company’s Board of Directors to its employees, directors and officers and to consultants, agents, advisors and independent contractors who provide services to the Company or to a subsidiary of the Company. The exercise price for stock options must not be less than the fair market value of the underlying shares on the date of grant. Stock options expire no later than ten years from the date of grant and generally vest and typically become exercisable over a four-year period following the date of grant. Under the 2016 Plan, the number of shares authorized for issuance automatically increases annually beginning January 1, 2017 and through January 1, 2026.
In connection with the Merger, the Company assumed the C3J Jian, Inc. Amended 2006 Stock Option Plan (the “Assumed 2006 Plan”) and the C3J Therapeutics, Inc. 2016 Stock Plan (the “Assumed 2016 Plan”). These plans provided for stock option and restricted stock awards (“RSAs”) to C3J employees in years prior to the merger with AmpliPhi. The number of shares subject to each outstanding stock option and RSA under those assumed plans, along with the exercise price of stock options, were equitably adjusted pursuant to the terms of the plans to reflect the impact of the Merger and the one-for-fourteen reverse stock split, in each case in a manner intended to preserved the then-current intrinsic value of the awards. No additional awards will be made under either plan. The assumed C3J stock options were substantially vested and expensed as of the merger date. Vesting of the assumed C3J RSAs is based on the occurrence of a public liquidity event, or a change in control. In the event of a public liquidity event, service or milestone based vesting schedules begins. Service periods are generally two to four years. In the event of a change in control, 100% vesting occurs upon the closing of such an event. The merger with AmpliPhi constituted a public liquidity event and triggered the start of vesting of RSAs.
Stock-based Compensation
The Company estimates the fair value of stock options with performance and service conditions using the Black-Scholes valuation model. Compensation expense related to stock options granted is measured at the grant date based on the estimated fair value of the award and is recognized on the accelerated attribution method over the requisite service period.
The assumptions used in the Black-Scholes model are presented below:
|
|
|
|
|
|
|
|
|
Three months ended
|
|
|
|
March 31, 2020
|
|
March 31, 2019
|
|
Risk-free interest rate
|
|
1.48% - 1.51%
|
|
—
|
|
Expected volatility
|
|
90.43%
|
|
—
|
|
Expected term (in years)
|
|
5.75 - 6.25
|
|
—
|
|
Expected dividend yield
|
|
0
|
|
0
|
The risk-free interest rate is based on the U.S. Treasury yield for a period consistent with the expected term of the option in effect at the time of the grant. Expected volatility is based on the historical volatility of Armata and peer companies’ common stock. The expected term represents the period that the Company expects its stock options to be outstanding. The expected term assumption is estimated using the simplified method set forth in the SEC Staff Accounting Bulletin 110, which is the mid-point between the option vesting date and the expiration date. For stock options granted to parties other than employees or directors, the Company elects, on a grant by grant basis, to use the expected term or the contractual term of the option award. The Company has never declared or paid dividends on its common stock and has no plans to do so in the foreseeable future. Forfeitures are recognized as a reduction of stock-based compensation expense as they occur.
The tables below summarize the total stock-based compensation expense included in the Company’s consolidated statements of operations for the periods presented:
|
|
|
|
|
|
|
|
|
|
|
|
Three Months Ended March 31,
|
|
|
|
|
2020
|
|
2019
|
|
|
|
|
|
|
|
|
|
|
|
Research and development
|
|
$
|
337,000
|
|
$
|
—
|
|
|
General and administrative
|
|
|
707,000
|
|
|
—
|
|
|
Total stock-based compensation
|
|
$
|
1,044,000
|
|
$
|
—
|
|
Stock option transactions during the three months ended March 31, 2020 are presented below:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options Outstanding
|
|
|
|
|
|
|
|
|
Weighted
|
|
|
|
|
|
|
|
|
|
|
|
Average
|
|
|
|
|
|
|
|
|
Weighted
|
|
Remaining
|
|
|
|
|
|
|
|
|
Average
|
|
Contractual
|
|
Aggregate
|
|
|
|
|
|
Exercise
|
|
Term
|
|
Intrinsic
|
|
|
|
Shares
|
|
Price
|
|
(Years)
|
|
Value
|
|
Outstanding at December 31, 2019
|
|
1,275,380
|
|
$
|
7.61
|
|
8.81
|
|
|
—
|
|
Granted
|
|
129,766
|
|
|
3.85
|
|
—
|
|
|
—
|
|
Forfeited/Cancelled
|
|
(39,382)
|
|
|
8.29
|
|
—
|
|
|
—
|
|
Outstanding at March 31, 2020
|
|
1,365,764
|
|
$
|
7.08
|
|
8.48
|
|
|
—
|
|
Vested and expected to vest at March 31, 2020
|
|
1,365,764
|
|
$
|
7.08
|
|
8.48
|
|
$
|
—
|
|
Exercisable at March 31, 2020
|
|
169,558
|
|
$
|
33.83
|
|
3.27
|
|
$
|
—
|
Restricted stock award transactions under the Assumed 2016 Plan during the three months ended March 31, 2020 are presented below:
|
|
|
|
|
|
|
|
|
|
|
|
Weighted Avg
|
|
|
|
|
|
Grant Date
|
|
|
|
Shares
|
|
Fair Value
|
|
Outstanding at December 31, 2019
|
|
343,493
|
|
$
|
21.83
|
|
Forfeited/Cancelled
|
|
(3,329)
|
|
|
16.14
|
|
Outstanding at March 31, 2020
|
|
340,164
|
|
$
|
21.91
|
The aggregate intrinsic value of options at March 31, 2020 is based on the Company’s closing stock price on that date of $3.10 per share. As of March 31, 2020, there was $4.8 million of total unrecognized compensation expense related to unvested stock options and RSAs, excluding unvested RSAs with performance factors deemed to be improbable for the period ending March 31, 2020, which the Company expects to recognize over the weighted average remaining period of approximately 2 years.
Shares Reserved for Future Issuance
As of March 31, 2020, the Company had reserved shares of its common stock for future issuance as follows:
|
|
|
|
|
|
|
Shares Reserved
|
|
Stock options outstanding
|
|
1,365,764
|
|
Employee stock purchase plan
|
|
7,605
|
|
Available for future grants under the 2016 Plan
|
|
469,433
|
|
Warrants outstanding
|
|
10,547,363
|
|
Total shares reserved
|
|
12,390,165
|
10. Commitments and Contingencies
The Company leases office and research and development space under a noncancelable ten-year operating lease in Marina Del Rey, CA. The lease commenced January 1, 2012 with the Company’s option to extend the lease for an additional ten years.
In April 2020, the Company entered into an Assignment and First Amendment of Office Lease (“Lease Amendment”) for its location in Marina Del Rey, which, among other things, extended the lease term for ten years commencing January 1, 2022 to expire on December 31, 2031. Base annual rent for calendar year 2022 under the Lease Amendment will be approximately $1.9 million, and base rent increases by 3% annually and will be $2.5 million by the end of the amended term. In addition, the Company received rent abatement for six months starting May 1, 2020, and allowance for tenant improvements of $0.8 million to be used during calendar year of 2021. The Company expects to remeasure the lease liability and related right of use asset upon Lease Amendment in connection with close of the consolidated financial statements for the six months ended June 30, 2020.
From time to time, the Company may be involved in disputes, including litigation, relating to claims arising out of operations in the normal course of business. Any of these claims could subject the Company to costly legal expenses and, while management generally believes that there is adequate insurance to cover many different types of liabilities, the Company’s insurance carriers may deny coverage or policy limits may be inadequate to fully satisfy any damage awards or settlements. If this were to happen, the payment of any such awards could have a material adverse effect on the consolidated results of operations and financial position. Additionally, any such claims, whether or not successful, could damage the Company’s reputation and business. The Company is currently not a party to any legal proceedings, the adverse outcome of which, in management’s opinion, individually or in the aggregate, would have a material adverse effect on our consolidated results of operations or financial position.
11. Cystic Fibrosis Foundation Award
On March 13, 2020, the Company entered into an award agreement (the “Agreement”) with Cystic Fibrosis Foundation (“CFF”), pursuant to which it received a development award of up to $5.0 million (the “Award”). The Award will be used to fund a portion of the Company’s Phase 1b/2 clinical trial of the Pseudomonas aeruginosa phage candidate, AP-PA02, as a treatment for Pseudomonas airway infections in people with cystic fibrosis (“CF”).
The first payment under the Agreement, in the amount of $1.0 million, became due upon signing the Agreement and was received in April 2020. The remainder of the Award will be paid to the Company incrementally in installments upon the achievement of certain milestones related to the development program and progress of the Phase 1b/2 clinical trial of AP-PA02, as set forth in the Agreement.
If the Company ceases to use commercially reasonable efforts directed to the development of AP-PA02, or any other Product (as defined in the Agreement), for a period of 360 days (an “Interruption”) and fails to resume the development of the Product after receiving from CFF notice of an Interruption, then the Company must either repay the amount of the Award actually received by the Company, plus interest, or grant to CFF (1) an exclusive (even as to the Company), worldwide, perpetual, sublicensable license under technology developed under the Agreement that covers the Product for use in treating infections in CF patients (the “CF Field”), and (2) a non-exclusive, worldwide, perpetual, sublicensable license under certain background intellectual property covering the Product, to the extent necessary to commercialize the Product in the CF Field.
Upon commercialization by the Company of any Product, the Company will owe a fixed royalty amount to CFF, which is to be paid in installments determined, in part, based on commercial sales volumes of the Product. The Company will be obligated to make an additional fixed royalty payment upon achieving specified sales milestones. The Company may also be obligated to make a payment to CFF if the Company transfers, sells or licenses the Product in the CF Field, or if the Company enters into a change of control transaction.
The term of the Agreement commenced on March 10, 2020 and expires on the earlier of the date on which the Company has paid CFF all of the fixed royalty payments set forth therein, the effective date of any license granted to
CFF following an Interruption, or upon earlier termination of the Agreement. Either CFF or the Company may terminate the agreement for cause, which includes the Company’s material failure to achieve certain development milestones. The Company’s payment obligations survive the termination of the Agreement.
The Company concluded that the CFF award is in the scope of ASC 808. Accordingly, as discussed in Note 3, award amounts received from CFF upon achievement of certain milestones are recognized as credits to research and development expenses in the period the expenses are incurred. In addition, the Company concluded under the guidance in ASC 730 that it does not have an obligation to repay funds received once related research and development expenses are incurred. Therefore, Armata recorded a liability for advances from CFF of $0.9 million as of March 31, 2020 representing amounts due to be received but not yet spent on the research program.
12. Synthetic Genomics Asset Acquisition
On February 28, 2018, C3J completed an acquisition of certain synthetic phage assets (the “synthetic phage assets”) from “SGI” for consideration consisting of $8.0 million in cash and $27.0 million in equity. The cash payments consisted of: $1.0 million paid at closing on February 28, 2018, $1.0 million at one year from closing, $1.0 million at two years from closing, and $5.0 million at three years from closing (the payments due on the one, two, three year anniversary are collectively the “time-based payment obligation”). The equity payment (the “equity payment” and, together with the time-based payment obligation, the “deferred purchase price arrangement”) is due upon the earlier of the initial public offering of shares of C3J’s common stock pursuant to an effective registration statement under the Securities Act of 1933, as amended, the sale of all or substantially all of C3J’s assets to a third party, or a consolidation or merger into a third party. On December 20, 2018, in contemplation of the Merger (see Note 5), the deferred purchase price arrangement was amended. Under the amended agreement, the purchase consideration consisted of (i) closing consideration of $1.0 million paid on February 28, 2018, (ii) cash payments of $1.0 million on January 31, 2019, $1.0 million on January 31, 2020, and $2.0 million on January 31, 2021, (iii) an issuance of that number of shares of C3J’s common stock equal to ten percent of C3J’s fully-diluted capitalization, excluding options and restricted stock awards, immediately prior to the closing of the Merger, and (iv) potential milestone payments of up to $39.5 million related to the development and relevant regulatory approval of products utilizing bacteriophage from the synthetic phage assets acquired from SGI (the “milestone payment obligation”).
The equity payment was determined to be a derivative liability in accordance with ASC 815, Derivatives and Hedging and was initially recorded at its fair value of $2.8 million. Throughout 2018 and until May 9, 2019, the derivative liability was adjusted to its fair value based upon a payment probability assessment and marked-to-market at the end of each period (see Note 4). Following the December 20, 2018 amendment to the deferred purchase price arrangement, the Company considered the probability of the reduction to the share issuance consideration in estimating the fair value of the derivative liability. For the three months periods ended March 31, 2019, the Company recognized $40,000 of interest expense related to the time-based payment obligations.
In connection with the Merger, the Company converted its equity payment obligation to SGI by issuing 516,976 shares of C3J’s common stock in connection with the amended agreement, after considering the Merger exchange ratio and reverse stock split in the manner described above. Through May 9, 2019, the derivative liability associated with the equity payment was updated for its estimated market value. Upon closing of the Merger, the fair value of the derivative liability was estimated at zero as the equity payment is no longer required to be made in the future. The change in fair value is reflected in other income.
13. Subsequent Events
As mentioned in Note 10 above, the Company entered into a Lease Amendment in April 2020.
In April 2020, the Company received loan proceeds of $717,000 (“PPP Loan”) under the Paycheck Protection Program (“PPP”). The PPP, established as part of the Coronavirus Aid, Relief and Economic Security Act, provides for loans to qualifying businesses for amounts up to 2.5 times the average monthly payroll expenses of the qualifying business, calculated as provided under the PPP. The PPP provides a mechanism for forgiveness of up to the full amount borrowed after eight weeks as long as the borrower uses the loan proceeds during the eight-week period after the loan origination
for eligible purposes, including payroll costs, certain benefits costs, rent and utilities costs or other permitted purposes, and maintains its payroll levels, subject to certain other requirements and limitations. The amount of loan forgiveness is subject to reduction, among other reasons, if the borrower terminates employees or reduces salaries during the eight-week period. The Company cannot provide any assurance that it will be eligible for loan forgiveness or that any amount of the PPP loan will ultimately be forgiven.
The PPP Loan is unsecured, evidenced by a promissory note (the “Note”) given by the Company as borrower through its bank, serving as the lender. The interest rate on the Note is 1.0% per annum. Payments of principal and interest are deferred for seven months from the date of the Note (the “Deferral Period”). Any unforgiven portion of the PPP Loan is payable over the two-year term, with payments deferred during the Deferral Period. The Company is permitted to prepay the Note at any time without payment of any premium.
Item 2. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our unaudited consolidated financial statements and related notes included in this Quarterly Report on Form 10-Q, our audited financial statements and notes thereto as of and for the year ended December 31, 2019 included in our Form 10-K filed on March 19, 2020 with the U.S. Securities and Exchange Commission (the “SEC”).
Our predecessor, C3 Jian, Inc., was incorporated under the laws of the state of California on November 4, 2005. On February 26, 2016, as part of a reorganization transaction, C3 Jian, Inc. merged with a wholly owned subsidiary of C3J Therapeutics, Inc. (“C3J”), and as part of this process, C3 Jian, Inc. was converted to a limited liability company organized under the laws of the State of California named C3 Jian, LLC. On May 9, 2019, C3J completed a reverse merger with AmpliPhi Biosciences Corporation, a bacteriophage development stage company (“AmpliPhi”), where Ceres Merger Sub, Inc., a wholly-owned subsidiary of AmpliPhi, merged with and into C3J (the “Merger”). Following the completion of the Merger, and a $10.0 million concurrent private placement financing, the former C3J shareholders owned approximately 76% of our common stock and the former AmpliPhi shareholders owned approximately 24% of our common stock.
Immediately prior to the Merger, AmpliPhi completed a 1-for-14 reverse stock split and changed its name to Armata Pharmaceuticals, Inc. Our common stock is traded on the NYSE American exchange under the symbol “ARMP.” We are headquartered in Marina Del Rey, CA, in a 35,000 square-foot research and development facility built for product development with capabilities spanning from bench to clinic. In addition to microbiology, synthetic biology, formulation, chemistry and analytical laboratories, the facility is equipped with two licensed GMP drug manufacturing suites enabling the production, testing and release of clinical material.
Statements contained in this report that are not statements of historical fact are forward-looking statements within the meaning of the U.S. Private Securities Litigation Reform Act of 1995. Such forward-looking statements include, without limitation, statements concerning product development plans, commercialization of our products, the expected market opportunity for our products, the use of bacteriophages and synthetic phages to kill bacterial pathogens, having resources sufficient to fund our operations into the second quarter of 2021, future funding sources, general and administrative expenses, clinical trial and other research and development expenses, costs of manufacturing, costs relating to our intellectual property, capital expenditures, the expected benefits of our targeted phage therapies strategy, the potential market for our products, tax credits and carry-forwards, and litigation-related matters. Words such as “believe,” “anticipate,” “plan,” “expect,” “intend,” “will,” “goal,” “potential” and similar expressions are intended to identify forward-looking statements, though not all forward-looking statements necessarily contain these identifying words. These statements are subject to risks and uncertainties. Our actual results could differ materially from those anticipated in these forward-looking statements as a result of various factors, including those set forth below under Part II, Item 1A, “Risk Factors” and elsewhere in this Quarterly Report on Form 10-Q. These forward-looking statements speak only as of the date on which they were made, and we undertake no obligation to update any forward-looking statements.
Overview
We are a clinical-stage biotechnology company focused on the development of precisely targeted bacteriophage therapeutics for the treatment of antibiotic-resistant infections using our proprietary bacteriophage-based technology. Bacteriophages or “phages” have a powerful and highly differentiated mechanism of action that enables binding to and killing specific bacteria, in contrast to traditional broad-spectrum antibiotics. We believe that phages represent a promising means to treat bacterial infections, especially those that have developed resistance to current standard of care therapies, including the so-called multidrug-resistant or “superbug” strains of bacteria. We are a leading developer of phage therapeutics which are uniquely positioned to address the growing worldwide threat of antibiotic-resistant bacterial infections.
We are combining our proprietary approach and expertise in identifying, characterizing and developing both naturally-occurring and engineered (synthetic) bacteriophages with our proprietary phage-specific GMP manufacturing capabilities to advance a broad pipeline of high-quality bacteriophage product candidates. We believe that synthetic phage, engineered using advances in sequencing and synthetic biology techniques, represent a promising means to advance phage therapy, including phage-based diagnostics and improving upon the ability of natural phage to treat bacterial infections, especially those that have developed resistance to current antibiotic therapies, including the multidrug-resistant or “superbug” bacterial pathogens.
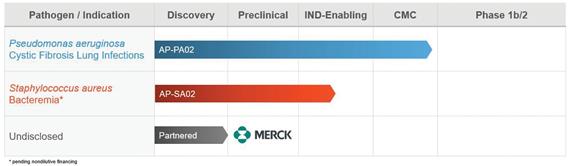
We are developing and advancing our second-generation phage product candidate for Pseudomonas aeruginosa (“P. aeruginosa”). We anticipate initiating a Phase 1b/2, multi-center, double-blind, randomized, placebo-controlled, single and multiple ascending dose study to evaluate the safety, tolerability, and preliminary efficacy in subjects with cystic fibrosis (“CF”) and chronic pulmonary P. aeruginosa infection in 2020, provided that COVID-19 disease has been reduced to the point that clinical trials in CF patients are enrolling. Prior to the COVID-19 pandemic, the Company had expected to initiate this clinical program in the first half of 2020.
We are also developing a second-generation phage product candidate for Staphylococcus aureus (“S. aureus”) for the treatment of S. aureus bacteremia. We intend to file an IND Application with the FDA to initiate a Phase 1/2, multi-center, randomized, double-blind, placebo- controlled dose escalation study that will assess the safety, tolerability, and efficacy of this development program in the second half of 2020 or the first half of 2021, depending on the timing of any third party non-dilutive financing that may be available toward funding of the program, as well as the impact of COVID-19 to our internal development efforts.
In partnership with Merck & Co., known as Merck Sharp & Dohme outside of the United States and Canada (“Merck”), we are developing proprietary synthetic phage candidates to target undisclosed infectious disease agents. Our proprietary phage engineering platform serves to enhance the clinical and commercial prospects of phage therapy. These attributes include expanded host range, improved potency which is a fundamental drug property that can translate into improved clinical efficacy, and importantly, biofilm disruption, which is a critical aspect of serious infections that needs to be addressed.
In addition to our more advanced pipeline programs, we have phage discovery efforts underway to target other major pathogens of infectious disease (including ESKAPE pathogens) and preventable infectious disease of the microbiome.
We are committed to conducting formal randomized clinical trials required for the Food and Drug Administration (“FDA”) approval in order to move toward commercialization of alternatives to traditional antibiotics and provide a potential method of treating patients suffering from drug-resistant bacterial infections.
The following chart summarizes the status of our phage product candidate development programs:
We have generally incurred net losses since our inception and our operations to date have been primarily limited to research and development and raising capital. As of March 31, 2020 we had an accumulated deficit of $162.6 million. We anticipate that a substantial portion of our capital resources and efforts in the foreseeable future will be focused on completing the development and seeking to obtain regulatory approval of our product candidates.
We currently expect to use our existing cash and cash equivalents for the continued research and development of our product candidates, including through our targeted phage therapies strategy, and for working capital and other general corporate purposes. We expect to continue to incur significant and increasing operating losses at least for the next several years. We do not expect to generate product revenue unless and until we successfully complete development and obtain marketing approval for at least one of our product candidates.
We may also use a portion of our existing cash and cash equivalents for the potential acquisition of, or investment in, product candidates, technologies, formulations or companies that complement our business, although we have no current understandings, commitments or agreements to do so. Our existing cash and cash equivalents will not be sufficient to enable us to complete all necessary development of any potential product candidates. Accordingly, we will be required to obtain further funding through one or more other public or private equity offerings, debt financings, collaboration, strategic financing, grants or government contract awards, licensing arrangements or other sources. Our ability to raise additional capital may be adversely impacted by potential worsening global economic conditions and the recent disruptions to, and volatility in, financial markets in the United States and worldwide resulting from the ongoing COVID-19 pandemic. Adequate additional funding may not be available to us on acceptable terms, or at all. If we are unable to raise capital when needed or on acceptable terms, we may be required to defer, reduce or eliminate significant planned expenditures, restructure, curtail or eliminate some or all of our development programs or other operations, dispose of assets, enter into arrangements that may require us to relinquish rights to certain of our product candidates, technologies or potential markets, file for bankruptcy or cease operations altogether. Any of these events could have a material adverse effect on our business, financial condition and results of operations and result in a loss of investment by our stockholders.
Recent Developments
In April 2020, we entered into an Assignment and First Amendment of Office Lease (“Lease Amendment”) for its location in Marina Del Rey, which, among other things, extended the lease term for ten years commencing January 1, 2022 to expire on December 31, 2031. Base annual rent for calendar year 2022 under the Lease Amendment will be approximately $1.9 million, and base rent increases by 3% annually and will be $2.5 million by the end of the amended term. In addition, we received rent abatement for six months starting May 1, 2020, and an allowance for tenant improvements of $0.8 million to be used during the calendar year of 2021. We expect to remeasure the lease liability and related right-of-use asset upon Lease Amendment in connection with close of the consolidated financial statements for the six months ended June 30, 2020.
On March 27, 2020, we completed a private placement transaction in which we sold to Innoviva a total of 8,710,800 newly issued shares of our common stock and warrants to purchase 8,710,800 shares of common stock, with an exercise
price per share of $2.87. The private placement was closed in two tranches for total aggregate gross proceeds of $25.0 million.
On January 30, 2020, the World Health Organization (“WHO”) announced a global health emergency because of a new strain of coronavirus originating in Wuhan, China (the “COVID-19 outbreak”) and the risks to the international community as the virus spreads globally beyond its point of origin. In March 2020, the WHO classified the COVID-19 outbreak as a pandemic, based on the rapid increase in exposure globally.
The full impact of the COVID-19 outbreak continues to evolve as of the date of this report. As such, it is uncertain as to the full magnitude that the pandemic will have on the Company’s financial condition, liquidity, and future results of operations. Management is actively monitoring the impact that the pandemic could have on its financial condition, liquidity, ability to enroll patients in its contemplated clinical trials, manufacturing and research and development operations, suppliers to our operations and suppliers to our outside clinical trial organizations, biotech industry overall, and importantly the health and safety of our workforce. Given the continuing evolution of the COVID-19 outbreak and the global responses to curb its spread, the Company is not able to estimate the effects of the COVID-19 outbreak on its results of operations, financial condition, or liquidity for fiscal year 2020.
In April 2020, the Company received loan proceeds of $717,000 (“PPP Loan”) under the Paycheck Protection Program (“PPP”). The PPP, established as part of the Coronavirus Aid, Relief and Economic Security Act, provides for loans to qualifying businesses for amounts up to 2.5 times the average monthly payroll expenses of the qualifying business, calculated as provided under the PPP. The PPP provides a mechanism for forgiveness of up to the full amount borrowed after eight weeks as long as the borrower uses the loan proceeds during the eight-week period after the loan origination for eligible purposes, including payroll costs, certain benefits costs, rent and utilities costs or other permitted purposes, and maintains its payroll levels, subject to certain other requirements and limitations. The amount of loan forgiveness is subject to reduction, among other reasons, if the borrower terminates employees or reduces salaries during the eight-week period. The Company cannot provide any assurance that it will be eligible for loan forgiveness or that any amount of the PPP loan will ultimately be forgiven.
The PPP Loan is unsecured, evidenced by a promissory note (the “Note”) given by the Company as borrower through its bank, serving as the lender. The interest rate on the Note is 1.0% per annum. Payments of principal and interest are deferred for seven months from the date of the Note (the “Deferral Period”). Any unforgiven portion of the PPP Loan is payable over the two-year term, with payments deferred during the Deferral Period. The Company is permitted to prepay the Note at any time without payment of any premium.
Results of Operations
As a result of the Merger, C3J was considered the accounting acquirer of AmpliPhi because C3J’s shareholders retained a majority control of ownership of the combined company subsequent to the Merger; therefore, the historical financial statements presented herein prior to the closing of the Merger are the historical financial statements of C3J.
Comparison of three months ended March 31, 2020 and 2019
Research and Development
Research and development expenses for the three months ended March 31, 2020 and 2019 were $2.8 million and $2.1 million, respectively. The net increase of $0.7 million was primarily related to a $0.3 million increase in stock-based compensation expense, $0.3 million increase in research and development consulting expenses, $0.1 million increase in personnel expenses, $0.1 million increase in lab supplies and consulting costs, and offset by $0.1 million contra research and development expenses from the CFF award.
General and Administrative
General and administrative expenses for the three months ended March 31, 2020 and 2019 were $2.2 million and $1.4 million, respectively. The net increase of $0.8 million was primarily due to a $0.7 million increase in stock-based
compensation expense, a $0.2 million increase in personnel-related expenses, a $0.2 million increase in insurance costs, and offset by a $0.3 million decrease in professional fees (legal, audit and investment banking) associated with the Merger.
Other Income (Expense)
For the three-month period ended March 31, 2020 and 2019, we recorded noncash interest expense of $0.2 million and $0.3 million, respectively, as a result of interest accretion on the time-based cash payments due in connection with the SGI asset acquisition. The $0.1 million decrease was primarily related to a reduced balance due to SGI as a result of cash payment of $1.0 million in January 2020.
Income Taxes
There was no income tax expense or benefit for the three months ended March 31, 2020 and 2019.
Operating activities
Net cash used in operating activities for the three months ended March 31, 2020 was $4.2 million, as compared to $2.5 million for the three months ended March 31, 2019. The increase of $1.7 million was due to a $1.3 million increase to net loss, $0.8 million increase in non-cash adjustments to cash used in operating activities, and a $1.2 million increase to cash used for operating assets and liabilities.
Investing activities
Net cash used in investing activities was $0.1 million for each of the three months ended March 31, 2020 and 2019. Cash used in investing activities was primarily due to capital equipment purchases.
Financing activities
Net cash provided by financing activities was $22.4 million for the three months ended March 31, 2020, which was primarily comprised of $23.3 million net proceeds raised from Innoviva Private Placement and $0.1 million proceeds received from warrant exercises, offset by a payment of $1.0 million in deferred consideration related to the time-based payment obligation in connection with the SGI asset acquisition. Net cash used for financing activities was $1.0 million for the three months ended March 31, 2019, which was comprised of a payment of $1.0 million in deferred consideration related to the time-based payment obligation in connection with the SGI asset acquisition.
Liquidity, Capital Resources and Financial Condition
We have prepared our consolidated financial statements on a going concern basis, which assumes that we will realize our assets and satisfy our liabilities in the normal course of business. However, we have incurred net losses since our inception and have negative operating cash flows. These circumstances raise substantial doubt about our ability to continue as a going concern. The accompanying financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classifications of liabilities that may result from the outcome of the uncertainty concerning our ability to continue as a going concern. While management believes this plan to raise additional funds will alleviate the conditions that raise substantial doubt, these plans are not entirely within its control and cannot be assessed as being probable of occurring. The Company may not be able to secure additional financing in a timely manner or on favorable terms, if at all.
As of March 31, 2020, we had unrestricted cash and cash equivalents of $24.2 million. Considering our current cash resources, management believes our existing resources will be sufficient to fund our planned operations into the second quarter of 2021. For the foreseeable future, our ability to continue its operations is dependent upon our ability to obtain additional capital.
Future Capital Requirements
We will need to raise additional capital in the future to continue to fund our operations. Our future funding requirements will depend on many factors, including:
|
·
| |
the costs and timing of our research and development activities; |
|
·
| |
the progress and cost of our clinical trials and other research and development activities; |
|
·
| |
manufacturing costs associated with our targeted phage therapies strategy and other research and development activities; |
|
·
| |
the terms and timing of any collaborative, licensing, acquisition or other arrangements that we may establish; |
|
·
| |
whether and when we receive future Australian tax rebates, if any; |
|
·
| |
the costs and timing of seeking regulatory approvals; |
|
·
| |
the costs of filing, prosecuting and enforcing any patent applications, claims, patents and other intellectual property rights; and |
|
·
| |
the costs of lawsuits involving us or our product candidates. |
We may seek to raise capital through a variety of sources, including:
|
·
| |
the public equity market; |
|
·
| |
private equity financings; |
|
·
| |
collaborative arrangements, government grants or strategic financings; |
|
·
| |
licensing arrangements; and |
|
·
| |
public or private debt. |
Any additional fundraising efforts may divert our management team from their day to day activities, which may adversely affect our ability to develop and commercialize our product candidates. Our ability to raise additional funds will depend, in part, on the success of our product development activities, including our targeted phage therapies strategy and any clinical trials we initiate, regulatory events, our ability to identify and enter into in-licensing or other strategic arrangements, and other events or conditions that may affect our value or prospects, as well as factors related to financial, economic and market conditions, many of which are beyond our control. We cannot be certain that sufficient funds will be available to us when required or on acceptable terms. If we are unable to secure additional funds on a timely basis or on acceptable terms, we may be required to defer, reduce or eliminate significant planned expenditures, restructure, curtail or eliminate some or all of our development programs or other operations, dispose of technology or assets, pursue an acquisition of our company by a third party at a price that may result in a loss on investment for our stockholders, enter into arrangements that may require us to relinquish rights to certain of our product candidates, technologies or potential markets, file for bankruptcy or cease operations altogether. Any of these events could have a material adverse effect on our business, financial condition and results of operations. Moreover, if we are unable to obtain additional funds on a timely basis, there will be substantial doubt about our ability to continue as a going concern and increased risk of insolvency and loss of investment by our stockholders. To the extent that additional capital is raised through the sale of equity or convertible debt securities, the issuance of such securities could result in dilution to our existing stockholders. Our ability to raise additional capital may be adversely impacted by potential worsening global
economic conditions and the recent disruptions to, and volatility in, financial markets in the United States and worldwide resulting from the ongoing COVID-19 pandemic.
Off-Balance Sheet Arrangements
As of March 31, 2020, we did not have off-balance sheet arrangements.
Recent Accounting Pronouncements
Refer to Note 3 of the condensed consolidated notes to the consolidated financial statements contained elsewhere in this report.
Item 3. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We are a smaller reporting company as defined by Rule 12b‑2 of the Securities Exchange Act of 1934, as amended, or the Exchange Act, and are not required to provide the information required under this item.
Item 4. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
We carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures as defined in Rules 13a‑15(e) and 15d‑15(e) under the Exchange Act, as of the end of the period covered by this quarterly report on Form 10-Q. In designing and evaluating the disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable and not absolute assurance of achieving the desired control objectives and management necessarily applies its judgment in evaluating the cost benefit relationship of possible controls and procedures. Based on this evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level as of March 31, 2020.
Changes in Internal Control over Financial Reporting
An evaluation was also performed under the supervision and with the participation of our management, including our Chief Executive Officer and our Chief Financial Officer, of any change in our internal control over financial reporting that occurred during our last fiscal quarter and that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting. That evaluation did not identify any change in our internal control over financial reporting that occurred during our latest fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
PART II. OTHER INFORMATION
Item 1. Legal Proceedings
In addition, from time to time, we are a party to certain litigation that is either judged to be not material or that arises in the ordinary course of business. We intend to vigorously defend our interests in these matters. We expect that the resolution of these matters will not have a material adverse effect on our business, financial condition or results of operations. However, due to the uncertainties inherent in litigation, no assurance can be given as to the outcome of these proceedings.
Item 1A. Risk Factors
You should consider carefully the following information about the risks described below, together with the other information contained in this Quarterly Report and in our other public filings in evaluating our business. If any of the
following risks actually occur, our business, financial condition, results of operations, and future growth prospects would likely be materially and adversely affected. In these circumstances, the market price of our common stock would likely decline.
Risks Related to Our Financial Condition and Need for Additional Capital
There is substantial doubt about our ability to continue as a going concern, which may affect our ability to obtain future financing and may require us to curtail our operations. We will need to raise additional capital to support our operations.
The audited financial statements and accompanying notes thereto as of and for the year ended December 31, 2019 included in the Company’s Form 10-K filed with the SEC on March 19, 2020, included disclosures and an opinion from our independent registered public accounting firm stating that our recurring losses and negative cash flows from operations raise substantial doubt about our ability to continue as a going concern. Our financial statements as of December 31, 2019 and March 31, 2020 were prepared under the assumption that we will continue as a going concern and do not include any adjustments that might result from the outcome of this uncertainty. At March 31, 2020, we had cash and cash equivalents of $24.2 million, and we have had recurring losses from operations and negative operating cash flows since inception.
We will need to raise additional capital to support our operations and product development activities. In the near term, we expect to continue to fund our operations, if at all, primarily through equity and debt financings in the future. Our ability to raise additional capital may be adversely impacted by potential worsening global economic conditions and the recent disruptions to, and volatility in, financial markets in the United States and worldwide resulting from the ongoing COVID-19 pandemic. We may also seek funds through arrangements with collaborators, grant agencies or others that may require us to relinquish rights to the product candidates that we might otherwise seek to develop or commercialize independently. If we are unable to secure additional funds when needed or on acceptable terms, we may be required to defer, reduce or eliminate significant planned expenditures, restructure, curtail or eliminate some or all of our development programs or other operations, dispose of technology or assets, pursue an acquisition of our company by a third party at a price that may result in a loss on investment for our stockholders, enter into arrangements that may require us to relinquish rights to certain of our product candidates, technologies or potential markets, file for bankruptcy or cease operations altogether. Any of these events could have a material adverse effect on our business, financial condition and results of operations.
On January 27, 2020, we entered into a Securities Purchase Agreement (the “Securities Purchase Agreement”) with Innoviva, Inc. (“Innoviva”) pursuant to which we sold 8,710,800 newly issued shares of common stock, par value $0.01 per share of Armata and warrants to purchase 8,710,800 shares of common stock, with an exercise price per share of $2.87, in exchange for aggregate gross proceeds of approximately $25.0 million.
While we believe that our existing resources will be sufficient to fund our planned operations into the second quarter of 2021, we cannot provide assurances that our estimates are accurate, that our plans will not change or that changed circumstances will not result in the depletion of our capital resources more rapidly than we currently anticipate. Developing drugs and conducting clinical trials is expensive. Our future funding requirements will depend on many factors, including:
|
·
| |
the costs and timing of our research and development activities; |
|
·
| |
the progress and cost of our clinical trials and other research and development activities; |
|
·
| |
manufacturing costs associated with our targeted phage therapies strategy and other research and development activities; |
|
·
| |
the terms and timing of any collaborative, licensing, acquisition or other arrangements that we may establish; |
|
·
| |
whether and when we receive future Australian tax rebates, if any; |
|
·
| |
the costs and timing of seeking regulatory approvals; |
|
·
| |
the costs of filing, prosecuting, defending and enforcing any patent applications, claims, patents and other intellectual property rights; and |
|
·
| |
the costs of lawsuits involving us or our product candidates. |
In addition, raising additional capital through the sale of securities could cause significant dilution to our stockholders. Any additional fundraising efforts may divert our management from their day to day activities, which may adversely affect our ability to develop and commercialize our product candidates. Our ability to raise additional funds will depend, in part, on the success of our product development activities, including our targeted phage therapies strategy and any clinical trials we initiate, regulatory events, our ability to identify and enter into in-licensing or other strategic arrangements, and other events or conditions that may affect our value or prospects, as well as factors related to financial, economic and market conditions, many of which are beyond our control. There can be no assurances that sufficient funds will be available to us when required or on acceptable terms, if at all.
We have incurred losses since our inception and anticipate that we will continue to incur significant losses for the foreseeable future, and our future profitability is uncertain.
As of March 31, 2020, our accumulated deficit was $162.6 million and we expect to incur losses for the foreseeable future. We have devoted, and will continue to devote for the foreseeable future, substantially all of our resources to research and development of our product candidates. For the three months ended March 31, 2020, we had loss from operations of $4.9 million. For the years ended December 31, 2019 and 2018, we had losses from operations of $19.8 million and $17.7 million, respectively. Additional information regarding our results of operations may be found in our consolidated financial statements and in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included in Item 2 in this report.
We have never generated any revenue from product sales and may never be profitable.
Clinical trials and activities associated with discovery research are costly. We do not expect to generate any revenue from the commercial sales of our product candidates in the near term, and we expect to continue to have significant losses for the foreseeable future.
Our ability to generate meaningful revenue and achieve profitability depends on successfully completing the development of, and obtaining the regulatory approvals necessary to, commercialize our product candidates. If any of our product candidates fail in clinical trials or if any of our product candidates do not gain regulatory approval, or if any of our product candidates, if approved, fail to achieve market acceptance, we may never become profitable. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods. Our ability to generate future revenues from product sales depends heavily on our success in:
|
·
| |
completing research and preclinical and clinical development of our product candidates; |
|
·
| |
seeking and obtaining regulatory and marketing approvals for product candidates for which we complete clinical trials; |
|
·
| |
developing a sustainable, scalable, reproducible, and transferable manufacturing process for our product candidates; |
|
·
| |
launching and commercializing product candidates for which we obtain regulatory and marketing approval, either by establishing a sales force, marketing and distribution infrastructure, or by collaborating with a partner; |
|
·
| |
obtaining market acceptance of any approved products; |
|
·
| |
addressing any competing technological and market developments; |
|
·
| |
implementing additional internal systems and infrastructure, as needed; |
|
·
| |
identifying and validating new product candidates; |
|
·
| |
negotiating favorable terms in any collaboration, licensing or other arrangements into which we may enter; |
|
·
| |
maintaining, protecting and expanding our portfolio of intellectual property rights, including patents, trade secrets and know-how; and |
|
·
| |
attracting, hiring and retaining qualified personnel. |
Even if one or more of the product candidates that we develop is approved for commercial sale, we anticipate incurring significant costs associated with commercializing any approved product. Our expenses could increase beyond expectations if we are required by the FDA, the European Medicines Agency (“EMA”), or other foreign regulatory authorities to perform clinical trials and other studies in addition to those that we currently anticipate. Even if we are able to generate revenues from the sale of any approved products, we may not become profitable and may need to obtain additional funding to continue operations.
The coronavirus outbreak could adversely affect our results of operations.
During January 2020, a strain of the COVID-19 viral disease (or the “coronavirus”) was reported to have surfaced in Wuhan, China. In an effort to halt the outbreak, the Chinese government placed significant restrictions on travel within China and closed certain businesses in the region, and governments and other parties outside of China have halted or sharply curtailed the movement of people, goods and services. In March 2020, the World Health Organization called the COVID-19 viral disease a pandemic and the United States has been substantially impacted by the outbreak including the closing of many businesses, work from home requirements for most U.S. workers and substantial government intervention into the US economy. If the impact of the coronavirus outbreak continues for an extended period, it could materially adversely impact our clinical development activities as a result of the impacts on our employees, our manufacturing facility, our supply of critical laboratory and manufacturing materials, our clinical trial sites, access to patients in the clinical trials and additionally regulatory guidance could be delayed or impacted. At this point, we cannot accurately predict what effects these conditions will have on our business, which will depend on, among other factors, the ultimate geographic spread of the virus, the duration of the outbreak, the timing and restrictions that will be placed on our operations that those that support our business when we do begin to return to more normal operations and travel restrictions and business closures imposed by the various governments.
The 2017 comprehensive tax reform bill could adversely affect our business and financial condition.
On December 22, 2017, President Trump signed into law new legislation that significantly revises the Internal Revenue Code of 1986, as amended. The newly enacted federal income tax law, among other things, contains significant changes to corporate taxation, including reduction of the corporate tax rate from a top marginal rate of 35% to a flat rate of 21%, limitation of the tax deduction for interest expense to 30% of adjusted earnings (except for certain small businesses), limitation of the deduction for net operating losses to 80% of current year taxable income and elimination of net operating loss carrybacks, one time taxation of offshore earnings at reduced rates regardless of whether they are repatriated, immediate deductions for certain new investments instead of deductions for depreciation expense over time, and modifying or repealing many business deductions and credits (including reducing the business tax credit for certain clinical testing expenses incurred in the testing of certain drugs for rare diseases or conditions). Notwithstanding the reduction in the corporate income tax rate, the overall impact of the new federal tax law is uncertain and our business and financial condition could be adversely affected. In addition, it is uncertain if and to what extent various states will conform to the newly enacted federal tax law.
Taxing authorities could reallocate our taxable income among our subsidiaries, which could increase our overall tax liability.
We are organized in the United States, and we currently have subsidiaries in the United Kingdom and Australia. If we succeed in growing our business, we expect to conduct increased operations through our subsidiaries in various tax jurisdictions pursuant to transfer pricing arrangements between us and our subsidiaries. If two or more affiliated companies are located in different countries, the tax laws or regulations of each country generally will require that transfer prices be the same as those between unrelated companies dealing at arm’s length and that appropriate documentation is maintained to support the transfer prices. While we believe that we operate in compliance with applicable transfer pricing laws and intend to continue to do so, our transfer pricing procedures are not binding on applicable tax authorities.
If tax authorities in any of these countries were to successfully challenge our transfer prices as not reflecting arm’s length transactions, they could require us to adjust our transfer prices and thereby reallocate our income to reflect these revised transfer prices, which could result in a higher tax liability to us. In addition, if the country from which the income is reallocated does not agree with the reallocation, both countries could tax the same income, resulting in double taxation. If tax authorities were to allocate income to a higher tax jurisdiction, subject our income to double taxation or assess interest and penalties, it would increase our consolidated tax liability, which could adversely affect our financial condition, results of operations and cash flows.
Maintaining and improving our financial controls and the requirements of being a public company may strain our resources, divert management’s attention and affect our ability to attract and retain qualified board members.
As a public company, we are subject to the reporting requirements of the Exchange Act, the Sarbanes-Oxley Act and the rules of the NYSE American. The requirements of these rules and regulations increase our legal and financial compliance costs, make some activities more difficult, time-consuming or costly and place strain on our personnel, systems and resources. The Exchange Act requires, among other things, that we file annual, quarterly and current reports with respect to our business and financial condition.
The Sarbanes-Oxley Act requires, among other things, that we maintain effective disclosure controls and procedures and internal control over financial reporting. Ensuring that we have adequate internal financial and accounting controls and procedures in place is a costly and time-consuming effort that needs to be re-evaluated frequently.
We currently do not have an internal audit group, and we may need to hire additional accounting and financial staff with appropriate public company experience and technical accounting knowledge. Implementing any appropriate changes to our internal controls may require specific compliance training for our directors, officers and employees, entail substantial costs to modify our existing accounting systems, and take a significant period of time to complete. Such changes may not, however, be effective in maintaining the adequacy of our internal controls, and any failure to maintain that adequacy, or consequent inability to produce accurate financial statements on a timely basis, could increase our operating costs and could materially impair our ability to operate our business. Moreover, effective internal controls are necessary for us to produce reliable financial reports and are important to help prevent fraud.
In accordance with NYSE American rules, we are required to maintain a majority independent board of directors. The various rules and regulations applicable to public companies make it more difficult and more expensive for us to maintain directors’ and officers’ liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to maintain coverage. If we are unable to maintain adequate directors’ and officers’ insurance, our ability to recruit and retain qualified officers and directors will be significantly curtailed.
If we fail to maintain proper and effective internal control over financial reporting, our ability to produce accurate financial statements on a timely basis could be impaired and our public reporting may be unreliable.
We are required to maintain internal control over financial reporting adequate to provide reasonable assurance regarding the reliability of financial reporting and the preparation of our consolidated financial statements in accordance with generally accepted accounting principles. We do not expect that our internal control over financial reporting will
prevent all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Controls can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the controls. Over time, controls may become inadequate because changes in conditions or deterioration in the degree of compliance with policies or procedures may occur. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected. Material weaknesses in our internal controls have been identified in the past, and we cannot assure you that significant deficiencies or material weaknesses in our internal control over financial reporting will not be identified in the future.
If we are unable to maintain effective controls and procedures, or identify any future material weaknesses, the accuracy and timing of our financial reporting may be adversely affected, we may be unable to maintain compliance with securities law requirements regarding timely filing of periodic reports and we may experience a loss of public confidence, which could have an adverse effect on our business, financial condition and the market price of our common stock.
Risks Related to Our Business
Because the Merger resulted in an ownership change under Section 382 of the Internal Revenue Code for Armata, Armata’s pre-Merger net operating loss carryforwards and certain other tax attributes will be subject to limitations. The net operating loss carryforwards and other tax attributes of C3J may also be subject to limitations as a result of ownership changes.
If a corporation undergoes an “ownership change” within the meaning of Section 382 of the Internal Revenue Code of 1986, as amended, such corporation’s net operating loss carryforwards and certain other tax attributes arising from before the ownership change are subject to limitations on use after the ownership change. In general, an ownership change occurs if there is a cumulative change in the corporation’s equity ownership by certain stockholders that exceeds fifty percentage points over a rolling three-year period. Similar rules may apply under state tax laws. The Merger resulted in an ownership change for AmpliPhi and, accordingly, AmpliPhi’s net operating loss carryforwards and certain other tax attributes may be subject to limitations (or disallowance) on their use after the Merger. C3J’s net operating loss carryforwards may also be subject to limitation as a result of prior shifts in equity ownership and/or the Merger. Additional ownership changes in the future could result in additional limitations on our net operating loss carryforwards. Consequently, even if we achieve profitability, we may not be able to utilize a material portion of our net operating loss carryforwards and other tax attributes, which could have a material adverse effect on cash flow and results of operations.
As of December 31, 2019, we had federal net operating loss carryforwards of approximately $88.8 million.
We have limited operating history, have incurred significant operating losses since inception and expects to incur significant operating losses for the foreseeable future. We may never become profitable or, if achieved, be able to sustain profitability.
To date, we have funded our operations primarily through private placement offerings of equity securities. As of March 31, 2020, we had cash and cash equivalents of $24.2 million. We have incurred significant operating losses since our inception and expect to incur significant losses for the foreseeable future as we continue our development programs for our product candidates.
We currently generate no revenue from product sales, and may never be able to commercialize our product candidates, or other future product candidates. We do not currently have the required approvals to market our product candidates and we may never receive them. We may not be profitable even if we or any of our future development partners succeed in commercializing any of our product candidates. Because of the numerous risks and uncertainties associated with developing and commercializing our product candidates, we are unable to predict the extent of any future losses or when it will become profitable, if at all.
Results from preclinical studies and Phase 1 or 2 clinical trials of our product candidates or from single-patient expanded access treatments may not be predictive of the results of later stage clinical trials.
Preclinical studies, including studies of our product candidates in animal disease models, may not accurately predict the result of human clinical trials of those product candidates. In particular, promising animal studies suggesting the efficacy of prototype phage products in the treatment of bacterial infections, such as P. aeruginosa and S. aureus, may not predict the ability of these products to treat similar infections in humans. Despite promising data in our completed Phase 1 clinical trials, our phage technology may be found not to be efficacious in treating bacterial infections alone or in combination with other agents, when studied in later-stage clinical trials.
In addition, we have used our bacteriophage technology in the area of targeted medicine under single-patient expanded access guidelines, which permit the use of phage therapy outside of clinical trials, in the United States and Australia. Despite prior single-patient expanded access successes, no assurance can be given that we will have similar single-patient expanded access treatment successes in the future. Single-patient expanded access is a term that is used to refer to the use of an investigational drug or therapy outside of a clinical trial to treat a patient with a serious or immediately life-threatening disease or condition who has no comparable or satisfactory alternative treatment options. Regulators often allow single-patient expanded access on a case-by-case basis for an individual patient or for defined groups of patients with similar treatment needs. In some countries, such as Australia, the treating physician can administer treatment under single-patient expanded access guidelines without pre-approval from the applicable regulatory authority.
In September 2018, we received the official minutes from our August 2018 Type B pre-IND meeting with the FDA regarding our AP-SA01 bacteriophage therapy product candidate. The FDA expressed general agreement with our proposed clinical trial designs and, based on the current FDA feedback, no additional clinical or nonclinical data are required to proceed with human clinical trials. Subsequent to the Type B meeting, we have changed the bacteriophage product candidate to AP-SA02. While we believe the FDA comments and stances related to AP-SA01 will apply to AP-SA02, there can be no assurances that is the case.
To satisfy FDA or foreign regulatory approval standards for the commercial sale of our product candidates, we must demonstrate in adequate and controlled clinical trials that our product candidates are safe and effective. Success in early clinical trials, including Phase 1 and Phase 2 trials, or in our single-patient expanded access program does not ensure that later clinical trials will be successful. Our initial results from early stage clinical trials or our single-patient expanded access program also may not be confirmed by later analysis or subsequent larger clinical trials. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials, even after obtaining promising results in earlier clinical trials and most product candidates that commence clinical trials are never approved for commercial sale.
We are seeking to develop antibacterial agents using bacteriophage and synthetic phage technology, a novel approach, which makes it difficult to predict the time and cost of development. No bacteriophage products have been approved in the United States or elsewhere.
We are developing our product candidates with bacteriophage and synthetic phage technology. We have not, nor to our knowledge has any other company, received regulatory approval from the FDA or equivalent foreign agencies for a pharmaceutical drug based on this approach. While in vitro studies have characterized the behavior of bacteriophages in cell cultures and there exists a body of literature regarding the use of phage therapy in humans, the safety and efficacy of phage therapy in humans has not been extensively studied in well-controlled modern clinical trials. Most of the prior research on phage-based therapy was conducted in the former Soviet Union prior to and immediately after World War II and lacked appropriate control group design or lacked control groups at all. Furthermore, the standard of care has changed substantially during the ensuing decades since those studies were performed, diminishing the relevance of prior claims of improved cure rates. We cannot be certain that our approach will lead to the development of approvable or marketable drugs.
Developing phage-based therapies on a commercial scale will also require developing new manufacturing processes and techniques. We and our third-party collaborators may experience delays in developing manufacturing capabilities for
our product candidates, and may not be able to do so at the scale required to efficiently conduct the clinical trials required to obtain regulatory approval of our product candidates, or to manufacture commercial quantities of our products, if approved.
In addition, the FDA or other regulatory agencies may lack experience in evaluating the safety and efficacy of drugs based on these approaches, which could lengthen the regulatory review process, increase our development costs and delay or prevent commercialization of our product candidates.
Delays in our clinical trials could result in us not achieving anticipated developmental milestones when expected, increased costs and delay our ability to obtain regulatory approval for and commercialize our product candidates.
Delays in our ability to commence or enroll patients for our clinical trials could result in us not meeting anticipated clinical milestones and could materially impact our product development costs and delay regulatory approval of our product candidates. Planned clinical trials may not be commenced or completed on schedule, or at all. Clinical trials can be delayed for a variety of reasons, including:
|
·
| |
delays in the development of manufacturing capabilities for our product candidates to enable their consistent production at clinical trial scale; |
|
·
| |
failures in our internal manufacturing operations that result in our inability to consistently and timely produce bacteriophages in sufficient quantities to support our clinical trials; |
|
·
| |
the availability of financial resources to commence and complete our planned clinical trials; |
|
·
| |
delays in reaching a consensus with clinical investigators on study design; |
|
·
| |
delays in reaching a consensus with regulatory agencies on trial design or in obtaining regulatory approval to commence a trial; |
|
·
| |
delays in obtaining clinical materials; |
|
·
| |
slower than expected patient recruitment for participation in clinical trials; |
|
·
| |
failure by clinical trial sites, other third parties, or us to adhere to clinical trial agreements; |
|
·
| |
delays in reaching agreement on acceptable clinical trial agreement terms with prospective sites or obtaining institutional review board approval; |
|
·
| |
changes in local regulations as part of a response to the COVID-19 outbreak, which may require us to change the ways in which our clinical trials are conducted, which may result in unexpected costs, or to discontinue the clinical trials altogether; and |
|
·
| |
adverse safety events experienced during our clinical trials. |
If we do not successfully commence or complete our clinical trials on schedule, the price of our common stock may decline.
Completion of clinical trials depends, among other things, on our ability to enroll a sufficient number of patients, which is a function of many factors, including:
|
·
| |
the therapeutic endpoints chosen for evaluation; |
|
·
| |
the eligibility criteria defined in the protocol; |
|
·
| |
the perceived benefit of the product candidate under study; |
|
·
| |
the size of the patient population required for analysis of the clinical trial’s therapeutic endpoints; |
|
·
| |
our ability to recruit clinical trial investigators and sites with the appropriate competencies and experience; |
|
·
| |
our ability to obtain and maintain patient consents; |
|
·
| |
delays or difficulties in enrolling patients in our clinical trials as a result of impacts associated with the COVID-19 pandemic; and |
|
·
| |
competition for patients from clinical trials for other treatments. |
We may experience difficulties in enrolling patients in our clinical trials, which could increase the costs or affect the timing or outcome of these clinical trials. This is particularly true with respect to diseases with relatively small patient populations.
We have not completed formulation development of our product candidates.
The development of our bacteriophage product candidates requires that we isolate, select and combine a number of bacteriophages that target the desired bacteria for that product candidate. The selection of bacteriophages for any of our product candidates is based on a variety of factors, including without limitation the ability of the selected phages, in combination, to successfully kill the targeted bacteria, the degree of cross-reactivity of the individual phages with the same part of the bacterial targets, the ability of the combined phages to satisfy regulatory requirements, our ability to manufacture sufficient quantities of the phages, intellectual property rights of third parties, and other factors. While we have selected initial formulations of AP-SA02 for the treatment of S. aureus infections, and the initial formulations of AP-PA02 for the treatment of Pseudomonas infections in CF patients. there can be no assurance that these initial formulations will be the final formulations of AP-SA02 or AP-PA02 for commercialization if approved. If we are unable to complete formulation development of our product candidates in the time frame that we have anticipated, then our product development timelines, and the regulatory approval of our product candidates, could be delayed.
Our product candidates must undergo rigorous clinical testing, such clinical testing may fail to demonstrate safety and efficacy and any of our product candidates could cause undesirable side effects, which would substantially delay or prevent regulatory approval or commercialization.
Before we can obtain regulatory approval for a product candidate, we must undertake extensive clinical testing in humans to demonstrate safety and efficacy to the satisfaction of the FDA or other regulatory agencies. Clinical trials of new drug candidates sufficient to obtain regulatory marketing approval are expensive and take years to complete.
We cannot be certain of successfully completing clinical testing within the time frame we have planned, or at all. We may experience numerous unforeseen events during, or as a result of, the clinical trial process that could delay or prevent us from receiving regulatory approval or commercializing our product candidates, including the following:
|
·
| |
our clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical and/or preclinical testing or to abandon programs; |
|
·
| |
the results obtained in earlier stage clinical testing may not be indicative of results in future clinical trials; |
|
·
| |
clinical trial results may not meet the level of statistical significance required by the FDA or other regulatory agencies; |
|
·
| |
we, or regulators, may suspend or terminate our clinical trials if the participating patients are being exposed to unacceptable health risks; and |
|
·
| |
our product candidates may have unintended or undesirable effects on patients that may delay or preclude regulatory approval of our product candidates or limit their commercial use, if approved. |
We must continue to develop manufacturing processes for our product candidates and any delay in or our inability to do so would result in delays in our clinical trials.
We are developing novel manufacturing processes for our product candidates at our facility in Marina Del Rey (near Los Angeles), California. The manufacturing processes for our product candidates, and the scale up of such processes for clinical trials, is novel, and there can be no assurance that we will be able to complete this work in a timely manner, if at all. Any delay in the development or scale up of these manufacturing processes could delay the start of clinical trials and harm our business. In the event our facility in Marina Del Rey does not receive a satisfactory current good manufacturing practice regulations (“cGMP”) inspection for the manufacture of our product candidates, we may need to fund additional modifications to our manufacturing process, conduct additional validation studies, or find alternative manufacturing facilities, any of which would result in significant cost to us as well as a delay of up to several years in obtaining approval for such product candidate.
Our manufacturing facility will be subject to ongoing periodic inspection by the FDA for compliance with U.S. cGMP regulations. Compliance with these regulations and standards is complex and costly, and there can be no assurance that we will be able to comply. Any failure to comply with applicable regulations could result in sanctions being imposed (including fines, injunctions and civil penalties), failure of regulatory authorities to grant marketing approval of our product candidates, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of product candidates or products, operating restrictions and criminal prosecution.
We may conduct clinical trials for our products or product candidates outside the United States and the FDA may not accept data from such trials.
We completed an investigator-sponsored clinical trial of AP-SA01 at the University of Adelaide in Australia for CRS in December 2016. Although the FDA may accept data from clinical trials conducted outside the United States, acceptance of such study data by the FDA is subject to certain conditions. For example, the study must be well designed and conducted and performed by qualified investigators in accordance with ethical principles. The study population must also adequately represent the U.S. population, and the data must be applicable to the U.S. population and U.S. medical practice in ways that the FDA deems clinically meaningful. Generally, the patient population for any clinical studies conducted outside of the United States must be representative of the population for whom we intend to label the product in the United States. In addition, such studies would be subject to the applicable local laws and FDA acceptance of the data would be dependent upon its determination that the studies also complied with all applicable U.S. laws and regulations. There can be no assurance the FDA will accept data from trials conducted outside of the United States. Further, with respect to AP-SA01, we have changed the product formulation to AP-SA02 and any work related to AP-SA01 may not be relevant to the FDA or other international regulatory authorities.
We may need to license additional intellectual property rights.
The development and commercialization of phage-based antibacterial agents may require us to obtain rights to intellectual property from third parties. We may also determine that it is necessary or advisable to license other intellectual property from third parties. There can be no assurance that such intellectual property rights would be available on commercially reasonable terms, if at all.
We are subject to significant regulatory approval requirements, which could delay, prevent or limit our ability to market our product candidates.
Our research and development activities, preclinical studies, clinical trials and the anticipated manufacturing and marketing of our product candidates are subject to extensive regulation by the FDA and other regulatory agencies in the United States and by comparable authorities in Europe and elsewhere. There can be no assurance that our manufacturing facilities will satisfy the requirements of the FDA or comparable foreign authorities. We require the approval of the relevant regulatory authorities before we may commence commercial sales of our product candidates in a given market.
The regulatory approval process is expensive and time-consuming, and the timing of receipt of regulatory approval is difficult to predict. Our product candidates could require a significantly longer time to gain regulatory approval than expected, or may never gain approval. We cannot be certain that, even after expending substantial time and financial resources, we will obtain regulatory approval for any of our product candidates. A delay or denial of regulatory approval could delay or prevent our ability to generate product revenues and to achieve profitability.
Changes in regulatory approval policies during the development period of any of our product candidates, changes in, or the enactment of, additional regulations or statutes, or changes in regulatory review practices for a submitted product application may cause a delay in obtaining approval or result in the rejection of an application for regulatory approval.
Regulatory approval, if obtained, may be made subject to limitations on the indicated uses for which we may market a product. These limitations could adversely affect our potential product revenues. Regulatory approval may also require costly post-marketing follow-up studies. In addition, the labeling, packaging, adverse event reporting, storage, advertising, promotion and record-keeping related to the product will be subject to extensive ongoing regulatory requirements. Furthermore, for any marketed product, its manufacturer and its manufacturing facilities will be subject to continual review and periodic inspections by the FDA or other regulatory authorities. Failure to comply with applicable regulatory requirements may, among other things, result in fines, suspensions of regulatory approvals, product recalls, product seizures, operating restrictions and criminal prosecution.
Failure to comply with health and data protection laws and regulations could lead to government enforcement actions (which could include civil or criminal penalties), private litigation and/or adverse publicity and could negatively affect our operating results and business.
We and any potential collaborators may be subject to federal, state and foreign data protection laws and regulations (i.e., laws and regulations that address privacy and data security). In the United States, numerous federal and state laws and regulations, including federal health information privacy laws, state data breach notification laws, state health information privacy laws and federal and state consumer protection laws (e.g., Section 5 of the Federal Trade Commission Act), that govern the collection, use, disclosure and protection of health-related and other personal information could apply to our operations or the operations of our collaborators. In addition, we may obtain health information from third parties (including research institutions from which we obtain clinical trial data) that are subject to privacy and security requirements under the HIPAA, as amended by HITECH. Depending on the facts and circumstances, we could be subject to criminal penalties if we knowingly obtain, use, or disclose individually identifiable health information maintained by a HIPAA-covered entity in a manner that is not authorized or permitted by HIPAA.
International data protection laws, including Regulation 2016/679, known as the General Data Protection Regulation (GDPR) may also apply to health-related and other personal information obtained outside of the United States. The GDPR went into effect on May 25, 2018. The GDPR introduced new data protection requirements in the European Union, as well as potential fines for noncompliant companies of up to the greater of €20 million or 4% of annual global revenue. The regulation imposes numerous new requirements for the collection, use and disclosure of personal information, including more stringent requirements relating to consent and the information that must be shared with data subjects about how their personal information is used, the obligation to notify regulators and affected individuals of personal data breaches, extensive new internal privacy governance obligations and obligations to honor expanded rights of individuals in relation to their personal information (e.g., the right to access, correct and delete their data). In addition, the GDPR includes restrictions on cross-border data transfer. The GDPR will increase our responsibility and liability in relation to personal data that we process, and we may be required to put in place additional mechanisms to ensure compliance with the new EU data protection rules. Further, the United Kingdom’s vote in favor of exiting the EU, often referred to as Brexit, has created uncertainty with regard to data protection regulation in the United Kingdom. In particular, it is unclear whether the United Kingdom will enact data protection legislation equivalent to the GDPR and how data transfers to and from the United Kingdom will be regulated.
In addition, California recently enacted legislation that has been dubbed the first “GDPR-like” law in the United States. Known as the California Consumer Privacy Act, it creates new individual privacy rights for consumers (as that word is broadly defined in the law) and places increased privacy and security obligations on entities handling personal data of consumers or households. When it goes into effect on January 1, 2020, the CCPA will require covered companies to provide new disclosures to California consumers, provide such consumers new ways to opt-out of certain
sales of personal information, and allow for a new cause of action for data breaches. Legislators have stated that amendments will be proposed to the CCPA before it goes into effect, but it remains unclear what, if any, modifications will be made to this legislation or how it will be interpreted. As currently written, the CCPA may impact (possibly significantly) our business activities and exemplifies the vulnerability of our business to the evolving regulatory environment related to personal data and protected health information.
Compliance with U.S. and international data protection laws and regulations could require us to take on more onerous obligations in our contracts, restrict our ability to collect, use and disclose data, or in some cases, impact our ability to operate in certain jurisdictions. Failure to comply with U.S. and international data protection laws and regulations could result in government enforcement actions (which could include civil or criminal penalties), private litigation and/or adverse publicity and could negatively affect our operating results and business. Moreover, clinical trial subjects about whom we or our potential collaborators obtain information, as well as the providers who share this information with us, may contractually limit our ability to use and disclose the information. Claims that we have violated individuals’ privacy rights, failed to comply with data protection laws, or breached our contractual obligations, even if we are not found liable, could be expensive and time consuming to defend and could result in adverse publicity that could harm our business.
A variety of risks associated with our international operations could materially adversely affect our business.
In addition to our U.S. operations, we have operations and subsidiaries in the United Kingdom and Australia. We face risks associated with our international operations, including possible unfavorable regulatory, pricing and reimbursement, political, tax and labor conditions, which could harm our business. We are subject to numerous risks associated with international business activities, including:
|
·
| |
compliance with differing or unexpected regulatory requirements for the development, manufacture and, if approved, commercialization of our product candidates; |
|
·
| |
difficulties in staffing and managing foreign operations; |
|
·
| |
foreign government taxes, regulations and permit requirements; |
|
·
| |
U.S. and foreign government tariffs, trade restrictions, price and exchange controls and other regulatory requirements; |
|
·
| |
anti-corruption laws, including the Foreign Corrupt Practices Act; |
|
·
| |
economic weakness, including inflation, natural disasters, war, events of terrorism or political instability in particular foreign countries; |
|
·
| |
fluctuations in currency exchange rates, which could result in increased operating expenses and reduced revenues, and other obligations related to doing business in another country; |
|
·
| |
compliance with tax, employment, immigration and labor laws, regulations and restrictions for employees living or traveling abroad; |
|
·
| |
workforce uncertainty in countries where labor unrest is more common than in the United States; |
|
·
| |
production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; |
|
·
| |
changes in diplomatic and trade relationships; and |
|
·
| |
challenges in enforcing our contractual and intellectual property rights, especially in those foreign countries that do not respect and protect intellectual property rights to the same extent as the United States. |
These and other risks associated with our international operations may materially adversely affect our business, financial condition and results of operations.
We do not have a sales force and do not currently have plans to develop one.
The commercial success of any of our product candidates will depend upon the strength of sales and marketing efforts for them. We do not have a sales force and have no experience in sales, marketing or distribution. To successfully commercialize our product candidates, we will need to develop such a capability ourselves or seek assistance from a third party with a large distribution system and a large direct sales force. We may be unable to put such a plan in place. In addition, if we arrange for others to market and sell our products, our revenues will depend upon the efforts of those parties. Such arrangements may not succeed. Even if one or more of our product candidates is approved for marketing, if we fail to establish adequate sales, marketing and distribution capabilities, independently or with others, our business will be materially harmed.
Our success depends in part on attracting, retaining and motivating our personnel.
Our success depends on our continued ability to attract, retain and motivate highly qualified management, clinical and scientific personnel and on our ability to develop and maintain important relationships with leading academic institutions, clinicians and scientists. Our success will depend on our ability to retain and motivate personnel and hire additional qualified personnel when required. Competition for qualified personnel in the biotechnology field is intense. We face competition for personnel from other biotechnology and pharmaceutical companies, universities, public and private research institutions and other organizations. We also face competition from other more well-funded and well-established businesses and we may also be viewed as a riskier choice from a job stability perspective due to our relative newer status than longer existing biotech and pharmaceutical companies. We may not be able to attract and retain qualified personnel on acceptable terms given the competition for such personnel. If we are unsuccessful in our retention, motivation and recruitment efforts, we may be unable to execute our business strategy.
We must manage a geographically dispersed organization.
While we are a small company, we currently have operations in the United States and Australia. In the future, we may also locate facilities in other locations based on proximity to personnel with the expertise needed to research, develop and manufacture phage-based therapeutics, costs of operations or other factors. Managing our organization across multiple locations and multiple time zones may reduce our efficiency, increase our expenses and increase the risk of operational difficulties in the execution of our plans.
Our business and operations might be adversely affected by security breaches, including any cybersecurity incidents.
We depend on the efficient and uninterrupted operation of our computer and communications systems, which we use for, among other things, sensitive company data, including our financial data, intellectual property and other proprietary business information.
While certain of our operations have business continuity and disaster recovery plans and other security measures intended to prevent and minimize the impact of IT-related interruptions, our IT infrastructure and the IT infrastructure of our consultants, contractors and vendors are vulnerable to damage from cyberattacks, computer viruses, unauthorized access, electrical failures and natural disasters or other catastrophic events. We could experience failures in our information systems and computer servers, which could result in an interruption of our normal business operations and require substantial expenditure of financial and administrative resources to remedy. System failures, accidents or security breaches can cause interruptions in our operations and can result in a material disruption of our targeted phage therapies, bacteriophage product candidates and other business operations. The loss of data from completed or future studies or clinical trials could result in delays in our research, development or regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were
to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liabilities and the development of our product candidates could be delayed or otherwise adversely affected.
Even though we believe we carry commercially reasonable business interruption and liability insurance, we might suffer losses as a result of business interruptions that exceed the coverage available under our insurance policies or for which we do not have coverage. For example, we are not insured against terrorist attacks or cyberattacks. Any natural disaster or catastrophic event could have a significant negative impact on our operations and financial results. Moreover, any such event could delay the development of our product candidates.
Interruptions in the availability of server systems or communications with Internet or cloud based services, or failure to maintain the security, confidentiality, accessibility or integrity of data stored on such systems, could harm our business.
We rely upon a variety of Internet service providers, third-party hosting facilities and cloud computing platform providers to support our business. Failure to maintain the security, confidentiality, accessibility or integrity of data stored on such systems could damage our reputation in the market, cause us to lose revenue or market share, increase our service costs, cause us to incur substantial costs, subject us to liability for damages and/or fines and divert our resources from other tasks, any one of which could materially adversely affect our business, financial condition, results of operations and prospects. Any damage to, or failure of, such systems, or communications to and between such systems, could result in interruptions in our operations. If our security measures or those of our third-party data center hosting facilities, cloud computing platform providers, or third-party service partners, are breached, and unauthorized access is obtained to our data or our information technology systems, we may incur significant legal and financial exposure and liabilities.
We do not have control over the operations of the facilities of our cloud service providers and our third-party providers may be vulnerable to damage or interruption from natural disasters, cybersecurity attacks, terrorist attacks, power outages and similar events or acts of misconduct. In addition, any changes in our cloud service providers’ service levels may adversely affect our ability to meet our requirements and operate our business.
Risks Related to Our Reliance on Third Parties
We will rely on third parties to conduct our clinical trials, and their failure to perform their obligations in a timely or competent manner may delay development and commercialization of our product candidates.
We expect to use third parties, such as clinical research organizations, to assist in conducting our clinical trials. However, we may face delays outside of our control if these parties do not perform their obligations in a timely or competent fashion or if we are forced to change service providers. This risk is heightened for clinical trials conducted outside of the United States, where it may be more difficult to ensure that clinical trials are conducted in compliance with FDA requirements. Any third party that we hire to conduct clinical trials may also provide services to our competitors, which could compromise the performance of their obligations to us. If we experience significant delays in the progress of our clinical trials and in our plans to submit Biologics License Applications, the commercial prospects for product candidates could be harmed and our ability to generate product revenue would be delayed or prevented.
Our clinical research operations could also be negatively impacted by delays resulting from the COVID-19 pandemic. We are not able to predict the impact on the timing and costs of our planned clinical trials as a result of COVID-19.
Risks Related to Our Intellectual Property
We are dependent on patents and proprietary technology. If we fail to adequately protect this intellectual property or if we otherwise do not have exclusivity for the marketing of our products, our ability to commercialize products could suffer.
Our commercial success will depend in part on our ability to obtain and maintain patent protection sufficient to prevent others from marketing our product candidates, as well as to defend and enforce these patents against infringement and to operate without infringing the proprietary rights of others. Protection of our product candidates from unauthorized use by third parties will depend on having valid and enforceable patents cover our product candidates or their manufacture or use, or having effective trade secret protection. If our patent applications do not result in issued patents, or if our patents are found to be invalid, we will lose the ability to exclude others from making, using or selling the inventions claimed therein. We have a limited number of patents and pending patent applications.
The patent positions of biotechnology companies can be uncertain and involve complex legal and factual questions. This is due to inconsistent application of policy and changes in policy relating to examination and enforcement of biotechnology patents to date on a global scale. The laws of some countries may not protect intellectual property rights to the same extent as the laws of countries having well-established patent systems, and those countries may lack adequate rules and procedures for defending our intellectual property rights. Also, changes in either patent laws or in interpretations of patent laws may diminish the value of our intellectual property. We are not able to guarantee that all of our patent applications will result in the issuance of patents and we cannot predict the breadth of claims that may be allowed in our patent applications or in the patent applications we may license from others.
Central provisions of The Leahy-Smith America Invents Act, or the America Invents Act went into effect on September 16, 2012 and on March 16, 2013. The America Invents Act includes a number of significant changes to U.S. patent law. These changes include provisions that affect the way patent applications are being filed, prosecuted and litigated. For example, the America Invents Act enacted proceedings involving post-issuance patent review procedures, such as inter partes review (“IPR”), and post-grant review, that allow third parties to challenge the validity of an issued patent in front of the United States Patent and Trademark Office (“U.S. PTO”) Patent Trial and Appeal Board. Each proceeding has different eligibility criteria and different patentability challenges that can be raised. IPRs permit any person (except a party who has been litigating the patent for more than a year) to challenge the validity of the patent on the grounds that it was anticipated or made obvious by prior art. Patents covering pharmaceutical products have been subject to attack in IPRs from generic drug companies and from hedge funds. If it is within six months of the issuance of the challenged patent, a third party can petition the U.S. PTO for post-grant review, which can be based on any invalidity grounds and is not limited to prior art patents or printed publications.
In post-issuance proceedings, U.S. PTO rules and regulations generally tend to favor patent challengers over patent owners. For example, unlike in district court litigation, claims challenged in post-issuance proceedings are given their broadest reasonable meaning, which increases the chance a claim might be invalidated by prior art or lack support in the patent specification. As another example, unlike in district court litigation, there is no presumption of validity for an issued patent, and thus, a challenger’s burden to prove invalidity is by a preponderance of the evidence, as opposed to the heightened clear and convincing evidence standard. As a result of these rules and others, statistics released by the U.S. PTO show a high percentage of claims being invalidated in post-issuance proceedings. Moreover, with few exceptions, there is no standing requirement to petition the U.S. PTO for inter partes review or post-grant review. In other words, companies that have not been charged with infringement or that lack commercial interest in the patented subject matter can still petition the U.S. PTO for review of an issued patent. Thus, even where we have issued patents, our rights under those patents may be challenged and ultimately not provide us with sufficient protection against competitive products or processes.
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
|
·
| |
we might not be the first to file patent applications for our inventions; |
|
·
| |
others may independently develop similar or alternative product candidates to any of our product candidates that fall outside the scope of our patents; |
|
·
| |
our pending patent applications may not result in issued patents; |
|
·
| |
our issued patents may not provide a basis for commercially viable products or may not provide us with any competitive advantages or may be challenged by third parties; |
|
·
| |
others may design around our patent claims to produce competitive products that fall outside the scope of our patents; |
|
·
| |
we may not develop additional patentable proprietary technologies related to our product candidates; and |
|
·
| |
we are dependent upon the diligence of our appointed agents in national jurisdictions, acting for and on our behalf, which control the prosecution of pending domestic and foreign patent applications and maintain granted domestic and foreign patents. |
An issued patent does not guarantee us the right to practice the patented technology or commercialize the patented product. Third parties may have blocking patents that could be used to prevent us from commercializing our patented products and practicing our patented technology. Our issued patents and those that may be issued in the future may be challenged, invalidated or circumvented, which could limit our ability to prevent competitors from marketing the same or related product candidates or could limit the length of the term of patent protection of our product candidates. Moreover, because of the extensive time required for development, testing and regulatory review of a potential product, it is possible that, before any of our product candidates can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of the patent. Patent term extensions may not be available for these patents.
We rely on trade secrets and other forms of non-patent intellectual property protection. If we are unable to protect our trade secrets, other companies may be able to compete more effectively against us.
We rely on trade secrets to protect certain aspects of our technology, including our proprietary processes for manufacturing and purifying bacteriophages. Trade secrets are difficult to protect, especially in the pharmaceutical industry, where much of the information about a product must be made public during the regulatory approval process. Although we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, outside scientific collaborators and other advisors may unintentionally or willfully disclose our information to competitors. Enforcing a claim that a third party illegally obtained and is using our trade secret information is expensive and time-consuming, and the outcome is unpredictable. In addition, courts outside the United States may be less willing to or may not protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how.
If we are sued for infringing intellectual property rights of third parties or if we are forced to engage in an interference proceeding, it will be costly and time-consuming, and an unfavorable outcome in that litigation or interference would have a material adverse effect on our business.
Our ability to commercialize our product candidates depends on our ability to develop, manufacture, market and sell our product candidates without infringing the proprietary rights of third parties. Numerous United States and foreign patents and patent applications, which are owned by third parties, exist in the general field of anti-infective products or in fields that otherwise may relate to our product candidates. If we are shown to infringe, we could be enjoined from use or sale of the claimed invention if we are unable to prove that the patent is invalid. In addition, because patent applications can take many years to issue, there may be currently pending patent applications, unknown to us, which may later result in issued patents that our product candidates may infringe, or which may trigger an interference proceeding regarding one of our owned or licensed patents or applications. There could also be existing patents of which
we are not aware that our product candidates may inadvertently infringe or which may become involved in an interference proceeding.
The biotechnology and pharmaceutical industries are characterized by the existence of a large number of patents and frequent litigation based on allegations of patent infringement. For so long as our product candidates are in clinical trials, we believe our clinical activities fall within the scope of the exemptions provided by 35 U.S.C. Section 271(e) in the United States, which exempts from patent infringement liability activities reasonably related to the development and submission of information to the FDA. As our clinical investigational drug product candidates progress toward commercialization, the possibility of a patent infringement claim against us increases. While we attempt to ensure that our active clinical investigational drugs and the methods we employ to manufacture them, as well as the methods for their use we intend to promote, do not infringe other parties’ patents and other proprietary rights, we cannot be certain they do not, and competitors or other parties may assert that we infringe their proprietary rights in any event.
We may be exposed to future litigation based on claims that our product candidates, or the methods we employ to manufacture them, or the uses for which we intend to promote them, infringe the intellectual property rights of others. Our ability to manufacture and commercialize our product candidates may depend on our ability to demonstrate that the manufacturing processes we employ and the use of our product candidates do not infringe third-party patents. If third-party patents were found to cover our product candidates or their use or manufacture, we could be required to pay damages or be enjoined and therefore unable to commercialize our product candidates, unless we obtained a license. A license may not be available to us on acceptable terms, if at all.
Risks Related to Our Industry
If our competitors are able to develop and market products that are more effective, safer or more affordable than ours, or obtain marketing approval before we do, our commercial opportunities may be limited.
Competition in the biotechnology and pharmaceutical industries is intense and continues to increase. Some companies that are larger and have significantly more resources than we do are aggressively pursuing antibacterial development programs, including traditional therapies and therapies with novel mechanisms of action. In addition, other companies are developing phage-based products for non-therapeutic uses, and may elect to use their expertise in phage development and manufacturing to try to develop products that would compete with ours.
We also face potential competition from academic institutions, government agencies and private and public research institutions engaged in the discovery and development of drugs and therapies. Many of our competitors have significantly greater financial resources and expertise in research and development, preclinical testing, conducting clinical trials, obtaining regulatory approvals, manufacturing, sales and marketing than we do. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established pharmaceutical companies.
Our competitors may succeed in developing products that are more effective, have fewer side effects and are safer or more affordable than our product candidates, which would render our product candidates less competitive or noncompetitive. These competitors also compete with us to recruit and retain qualified scientific and management personnel, establish clinical trial sites and patient registration for clinical trials, as well as to acquire technologies and technology licenses complementary to our programs or advantageous to our business. Moreover, competitors that are able to achieve patent protection, obtain regulatory approvals and commence commercial sales of their products before we do, and competitors that have already done so, may enjoy a significant competitive advantage.
The Generating Antibiotics Incentives Now Act is intended to provide incentives for the development of new, qualified infectious disease products. These incentives may result in more competition in the market for new antibiotics, and may cause pharmaceutical and biotechnology companies with more resources than we have to shift their efforts towards the development of products that could be competitive with our product candidates.
There is a substantial risk of product liability claims in our business. If we do not obtain sufficient liability insurance, a product liability claim could result in substantial liabilities.
Our business exposes us to significant potential product liability risks that are inherent in the development, manufacturing and marketing of human therapeutic products. Regardless of merit or eventual outcome, product liability claims may result in:
|
·
| |
delay or failure to complete our clinical trials; |
|
·
| |
withdrawal of clinical trial participants; |
|
·
| |
decreased demand for our product candidates; |
|
·
| |
injury to our reputation; |
|
·
| |
substantial monetary awards against us; and |
|
·
| |
diversion of management or other resources from key aspects of our operations. |
If we succeed in marketing products, product liability claims could result in an FDA investigation of the safety or efficacy of our products, our manufacturing processes and facilities or our marketing programs. An FDA investigation could also potentially lead to a recall of our products or more serious enforcement actions, or limitations on the indications, for which they may be used, or suspension or withdrawal of approval.
We have product liability insurance that covers our clinical trials up to a $10.0 million annual per claim and aggregate limit. We intend to expand our insurance coverage to include the sale of commercial products if marketing approval is obtained for our product candidates or any other compound that we may develop. However, insurance coverage is expensive and we may not be able to maintain insurance coverage at a reasonable cost or at all, and the insurance coverage that we obtain may not be adequate to cover potential claims or losses.
Even if we receive regulatory approval to market our product candidates, the market may not be receptive to our product candidates upon their commercial introduction, which would negatively affect our ability to achieve profitability.
Our product candidates may not gain market acceptance among physicians, patients, healthcare payors and the medical community. The degree of market acceptance of any approved products will depend on a number of factors, including:
|
·
| |
the effectiveness of the product; |
|
·
| |
the prevalence and severity of any side effects; |
|
·
| |
potential advantages or disadvantages over alternative treatments; |
|
·
| |
relative convenience and ease of administration; |
|
·
| |
the strength of marketing and distribution support; |
|
·
| |
the price of the product, both in absolute terms and relative to alternative treatments; and |
|
·
| |
sufficient third-party coverage or reimbursement. |
If our product candidates receive regulatory approval but do not achieve an adequate level of acceptance by physicians, healthcare payors and patients, we may not generate product revenues sufficient to attain profitability.
Foreign governments tend to impose strict price controls, which may adversely affect our future profitability.
In some foreign countries, particularly in the European Union, prescription drug pricing is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidate to other available therapies. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our profitability will be negatively affected.
We may incur significant costs complying with environmental laws and regulations, and failure to comply with these laws and regulations could expose us to significant liabilities.
Our research and development activities use biological and hazardous materials that are dangerous to human health and safety or the environment. We are subject to a variety of federal, state and local laws and regulations governing the use, generation, manufacture, storage, handling and disposal of these materials and wastes resulting from these materials. We are also subject to regulation by the Occupational Safety and Health Administration (“OSHA”), state and federal environmental protection agencies and to regulation under the Toxic Substances Control Act. OSHA, state governments or federal Environmental Protection Agency, may adopt regulations that may affect our research and development programs. We are unable to predict whether any agency will adopt any regulations that could have a material adverse effect on our operations. We have incurred, and will continue to incur, capital and operating expenditures and other costs in the ordinary course of our business in complying with these laws and regulations.
Although we believe our safety procedures for handling and disposing of these materials comply with federal, state and local laws and regulations, we cannot entirely eliminate the risk of accidental injury or contamination from the use, storage, handling or disposal of hazardous materials. In the event of contamination or injury, we could be held liable for any resulting damages, and any liability could significantly exceed our insurance coverage.
The uncertainty associated with pharmaceutical reimbursement and related matters may adversely affect our business.
Market acceptance and sales of any one or more of our product candidates will depend on reimbursement policies and may be affected by future healthcare reform measures in the United States and in foreign jurisdictions. Government authorities and third-party payers, such as private health insurers and health maintenance organizations, decide which drugs they will cover and establish payment levels. We cannot be certain that reimbursement will be available for any of our product candidates. Also, we cannot be certain that reimbursement policies will not reduce the demand for, or the price paid for, our products. If reimbursement is not available or is available on a limited basis, we may not be able to successfully commercialize any product candidates that we develop.
In the United States, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (the “MMA”), changed the way Medicare covers and pays for pharmaceutical products. The legislation established Medicare Part D, which expanded Medicare coverage for outpatient prescription drug purchases by the elderly but provided authority for limiting the number of drugs that will be covered in any therapeutic class. The MMA also introduced a new reimbursement methodology based on average sales prices for physician-administered drugs.
The United States and several foreign jurisdictions are considering, or have already enacted, a number of legislative and regulatory proposals to change the healthcare system in ways that could affect its ability to sell its products profitably. Among policy makers and payers in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access to healthcare. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives. we expect to experience pricing pressures in connection with
the sale of any products that we develop due to the trend toward managed healthcare, the increasing influence of health maintenance organizations, and additional legislative proposals.
In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (collectively, the “ACA”), became law in the United States, which substantially changed the way healthcare is financed by both governmental and private insurers. While we cannot predict what impact on federal reimbursement policies this legislation will have in general or on our business specifically, the ACA and any amendments thereto may result in downward pressure on pharmaceutical reimbursement, which could negatively affect market acceptance of, and the price we may charge for, our products that receive regulatory approval. We also cannot predict the impact of ACA and its amendments on us as many of the ACA, as amended, requires the promulgation of detailed regulations implementing the statutory provisions, which have not yet been fully implemented.
Risks Related to Our Common Stock
Innoviva may exert a substantial influence on actions requiring stockholder vote, potentially in a manner that you do not support.
Innoviva holds approximately 46.7% of our shares outstanding and accordingly controls approximately 46.7% of our voting power. In addition, Innoviva holds 8,710,800 warrants to purchase shares of our common stock following the second closing of the financing. If Innoviva were to exercise the warrants held by them, they would hold approximately 63.7% of our issued and outstanding shares of common stock. Innoviva’s large ownership stake may allow it to exert a substantial influence on actions requiring a stockholder vote, potentially in a manner that you do not support, including amendments to our articles of incorporation, election of our board of directors, removal of any of our directors, adoption of measures that could delay or prevent a change in control or impede a merger, takeover, or other business combination involving us, and approval of other major corporate transactions. In addition, Innoviva’s stock ownership may discourage a potential acquirer from making a tender offer or otherwise attempting to obtain control of us, which in turn could reduce our stock price or prevent our stockholders from realizing a premium over our stock price. Accordingly, our stockholders other than Innoviva may be unable to influence management and exercise control over our business.
The price of our common stock has been and may continue to be volatile.
As of March 31, 2020, we had outstanding common warrants to purchase an aggregate of 10,547,363 shares of our common stock at a weighted-average exercise price of $4.51 per share. As of March 31, 2020, in-the-money warrants included warrants issued to Innoviva during Private Placement which have an exercise price of $2.87 per share. We also have outstanding options to exercise 1,365,764 shares of our common stock at a weighted-average exercise price of $7.08 per share. Although we cannot determine when these warrants or options will ultimately be exercised, it is reasonable to assume that such warrants and options will be exercised only if the exercise price is below the market price of our common stock. To the extent any of our outstanding warrants or options are exercised, additional shares of our common stock will be issued that will generally be eligible for resale in the public market (subject to limitations under Rule 144 under the Securities Act for certain of our warrants and with respect to shares held by our affiliates), which will result in dilution to our security holders. The issuance of additional securities could also have an adverse effect on the market price of our common stock.
Provisions of Washington law and our current articles of incorporation and bylaws may discourage another company from acquiring us and may prevent attempts by our stockholders to replace or remove our current management.
Provisions of Washington law and our current articles of incorporation and bylaws may discourage, delay or prevent a merger or acquisition that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace or remove our board of directors. These provisions include:
|
·
| |
authorizing the issuance of “blank check” preferred stock without any need for action by stockholders; |
|
·
| |
requiring supermajority stockholder voting to effect certain amendments to our articles of incorporation and bylaws; and |
|
·
| |
establishing advance notice requirements for nominations for election to our board of directors or for proposing matters that can be acted on by stockholders at stockholder meetings. |
In addition, because we are incorporated in Washington, we are governed by the provisions of Chapter 23B.19 of the Washington Business Corporation Act, which, among other things, restricts the ability of stockholders owning 10% or more of our outstanding voting stock from merging or combining with us. These provisions could discourage potential acquisition attempts and could reduce the price that investors might be willing to pay for shares of our common stock in the future and result in the market price being lower than it would without these provisions.
Although we believe these provisions collectively provide for an opportunity to receive higher bids by requiring potential acquirers to negotiate with our board of directors, they would apply even if an offer may be considered beneficial by some stockholders. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it difficult for stockholders to replace members of our board of directors, which is responsible for appointing the members of our management
We have never paid dividends on our common stock, and we do not anticipate paying any cash dividends on our common stock in the foreseeable future.
We have never declared or paid cash dividends on our common stock. We do not anticipate paying any cash dividends on our common stock in the foreseeable future. We currently intend to retain all available funds and any future earnings to fund the development and growth of our business. As a result, capital appreciation, if any, of our common stock will be our stockholders’ sole source of gain for the foreseeable future.
If securities or industry analysts do not publish research or publish unfavorable research about our business, our stock price and trading volume could decline.
The trading market for our common stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. We currently have three securities analysts and may never obtain additional research coverage by other securities and industry analysts. If no additional securities or industry analysts commence coverage of our company, the trading price for our stock could be negatively impacted. If we obtain additional securities or industry analyst coverage and if one or more of the analysts who covers us downgrades our stock or publishes inaccurate or unfavorable research about our business, our stock price would likely decline. If one or more of these analysts ceases coverage of us or fails to publish reports on us regularly, demand for our stock could decrease, which could cause our stock price and trading volume to decline.
Sales of a substantial number of shares of our common stock in the public market by our existing stockholders could cause our stock price to decline.
Sales of a substantial number of shares of our common stock in the public market or the perception that these sales might occur, could depress the market price of our common stock and could impair our ability to raise capital through the sale of additional equity securities. We are unable to predict the effect that sales may have on the prevailing market price of our common stock.
Future sales and issuances of our common stock or rights to purchase common stock by us, including pursuant to our equity incentive plans, could result in additional dilution of the percentage ownership of our stockholders and could cause our stock price to decline.
We are generally not restricted from issuing additional common stock, including any securities that are convertible into or exchangeable for, or that represent the right to receive, common stock. The market price of our common stock could decline as a result of sales of common stock or securities that are convertible into or exchangeable for, or that represent the right to receive, common stock or the perception that such sales could occur.
We expect that significant additional capital will be needed in the future to continue our planned operations, including conducting clinical trials, commercialization efforts, expanded research and development activities and costs associated with operating as a public company. To the extent we raise additional capital by issuing equity or convertible securities, our stockholders may experience substantial dilution. We may sell common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell common stock, convertible securities or other equity securities in more than one transaction, investors may be materially diluted by subsequent sales. Such sales may also result in material dilution to our existing stockholders, and new investors could gain rights superior to our existing stockholders.
Pursuant to our 2016 Equity Incentive Plan (the “2016 Plan”), our management is authorized to grant stock options and other equity-based awards to our employees, directors and consultants. The number of shares available for future grant under the 2016 Plan will automatically increase on January 1st of each year by up to 5% of all shares of our capital stock outstanding as of December 31st of the preceding calendar year, subject to the ability of our board of directors to take action to reduce the size of the increase in any given year. In addition, we may grant or provide for the grant of rights to purchase shares of our common stock pursuant to our 2016 Employee Stock Purchase Plan (“ESPP”). The number of shares of our common stock reserved for issuance under the ESPP will automatically increase on January 1st of each calendar year by the lessor of 1% of the total number of shares of our common stock outstanding on December 31st of the preceding calendar year and 30,000 shares, subject to the ability of our board of directors to take action to reduce the size of the increase in any given year. Currently, we plan to register the increased number of shares available for issuance under the 2016 Plan and ESPP each year. Increases in the number of shares available for future grant or purchase may result in additional dilution, which could cause our stock price to decline.
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds
On March 27, 2020, we completed a private placement transaction in which we sold to Innoviva a total of 8,710,800 newly issued shares of the Company’s common stock and warrants to purchase 8,710,800 shares of common stock, with an exercise price per share of $2.87. The private placement was closed in two tranches for total aggregate gross proceeds of $25.0 million.
Registration Rights Agreement and Investor Rights Agreement
As part of the First Closing of the Private Placement, the Company entered into a registration rights agreement (the “Registration Rights Agreement”) and an investor rights agreement (the “Investor Rights Agreement”) with Innoviva. Pursuant to the Registration Rights Agreement, the Company must file a registration statement on Form S-1 or Form S-3 (the “Shelf Registration Statement”) covering the resale of the securities issued and sold pursuant to the Securities Purchase Agreement with the U.S. Securities and Exchange Commission (the “Commission”) on a continuous basis pursuant to Rule 415 promulgated under the Securities Act of 1933, as amended (the “Securities Act”), or if Rule 415 is not available for offers and sales of such securities, by such other means of distribution of such securities as Innoviva may reasonably specify. On April 8, 2020, the Shelf Registration Statement was declared effective under the Securities Act.
The Investor Rights Agreement provides that for so long as Innoviva and its affiliates hold at least 12.5% of the outstanding shares of Common Stock on a fully-diluted basis, Innoviva shall have the right to designate two (2) directors to the board of directors of the Company (the “Board”), and for so long as Innoviva and its affiliates hold at least 8% but less than 12.5% of the outstanding shares of Common Stock on a fully-diluted basis, Innoviva shall have the right to designate one (1) director to the Board, subject to certain qualifications and conditions in the Investor Rights Agreement. The Investor Rights Agreement also provides for participation rights for Innoviva to participate in future offerings of equity securities by the Company.
Item 3. Defaults upon Senior Securities
None.
Item 4. Mine Safety Disclosures
Not applicable.
Item 5. Other Information
None.
Item 6. Exhibits
|
Number
|
|
Description
|
|
|
|
|
|
3.1
|
|
Articles of Amendment to Articles of Incorporation of the Company (effective March 26, 2020) (incorporated herein by reference to Exhibit 3.1 to the Current Report on Form 8-K, filed with the SEC on March 30, 2020).
|
|
|
|
|
|
3.2
|
|
Amendment to Amended and Restated Bylaws of the Company (effective February 24, 2020) (incorporated herein by reference to Exhibit 3.1 to the Current Report on Form 8-K, filed with the SEC on February 26, 2020).
|
|
|
|
|
|
4.1
|
|
Form of Common Stock Warrant (incorporated herein by reference to Exhibit 4.1 to the Current Report on Form 8-K, filed with the SEC on January 29, 2020).
|
|
|
|
|
|
4.2
|
|
Registration Rights Agreement, dated February 12, 2020, by and between the Company and Innoviva (incorporated herein by reference to Exhibit 4.1 to the Current Report on Form 8-K, filed with the SEC on February 13, 2020).
|
|
|
|
|
|
10.1*
|
|
Letter Agreement, dated as of March 10, 2020, by and between Armata Pharmaceuticals, Inc. and the Cystic Fibrosis Foundation.
|
|
|
|
|
|
10.2
|
|
Employment Agreement, dated as of January 18, 2012, by and between C3 Jian, Inc. and Brian Varnum.
|
|
|
|
|
|
10.3
|
|
Assignment and First Amendment of Office Lease, dated as of April 2020, by and among Armata Pharmaceuticals, Inc., C3 Jian, Inc. and Marina Business Center, LLC.
|
|
|
|
|
|
10.4
|
|
Securities Purchase Agreement, dated January 27, 2020, by and between the Company and Innoviva (incorporated herein by reference to Exhibit 10.1 to the Current Report on Form 8-K, filed with the SEC on January 29, 2020).
|
|
|
|
|
|
10.5
|
|
Investor Rights Agreement, dated February 12, 2020, by and between the Company and Innoviva (incorporated herein by reference to Exhibit 10.1 to the Current Report on Form 8-K, filed with the SEC on February 13, 2020).
|
|
|
|
|
|
10.6
|
|
Form of Voting Agreement by and between Innoviva and certain stockholders of the Company (incorporated herein by reference to Exhibit 10.3 to the Current Report on Form 8-K, filed with the SEC on January 29, 2020).
|
|
|
|
|
|
31.1
|
|
Certification of Principal Executive Officer required by Rule 13a‑14(a) or Rule 15d‑14(a).
|
|
|
|
|
|
31.2
|
|
Certification of Principal Financial Officer required by Rule 13a‑14(a) or Rule 15d‑14(a).
|
|
|
|
|
|
32.1
|
|
Certification of Principal Executive Officer Required by Rule 13a‑14(b) or Rule 15d‑14(b) and 18 U.S.C. 1350.
|
|
|
|
|
* Indicates that certain identified information in the exhibit has been omitted because it is both (i) not material, and (ii) would likely cause competitive harm if publicly disclosed.
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
|
|
|
|
|
|
|
ARMATA PHARMACEUTICALS, INC.
|
|
|
|
|
Date: May 14, 2020
|
|
By
|
/s/ Todd R. Patrick
|
|
|
|
|
Name: Todd R. Patrick
|
|
|
|
|
Title: Chief Executive Officer
|
|
|
|
|
(Principal Executive Officer)
|
|
|
|
|
|
|
|
|
By
|
/s/ Steve R. Martin
|
|
|
|
|
Name: Steve R. Martin
|
|
|
|
|
Title: Chief Financial Officer
|
|
|
|
|
(Principal Financial and Accounting Officer)
|